1.本发明属于麻黄汤检测技术领域,尤其是一种麻黄汤指纹图谱检测方法。
背景技术:
2.麻黄汤,为解表剂,具有发汗解表,宣肺平喘之功效。主治外感风寒表实证。恶寒发热,头身疼痛,无汗而喘,舌苔薄白,脉浮紧。临床常用于治疗感冒、流行性感冒、急性支气管炎、支气管哮喘等属风寒表实证者。
3.指纹图谱检测是中药药剂的常用检测手段,可以保证中药药剂的安全、有效,保证药剂的品质稳定,同时有利于对中药作用机理进行深入研究,具体又包括薄层色谱法、高效液相色谱法、气相色谱法、光谱法等。
4.指纹图谱检测虽然是非常成熟的中药检测技术,但是不同的中药需要采用不同的检测参数,如何确定检测参数(如波长、流速、柱温、溶剂)是检测的难点。目前,部分学者作了麻黄汤指纹图谱检测的相关研究,但是并没有将检测参数标准化,在大批量生产麻黄汤时,需要严格、有效地检测标准。如cn202110600880与cn202110761770虽然公开了色谱定量测试的方法,但是采用的溶剂、流动相体系、流动相体系梯度等均未达到最优。
技术实现要素:
5.本发明所要解决的技术问题是提供一种麻黄汤指纹图谱检测方法,可实现检测的标准化,以用于批量化生产。
6.为解决上述问题,本发明采用的技术方案为:麻黄汤指纹图谱检测方法,包括
7.s1、制备麻黄汤煎液,将煎液浓缩至密度为1.2至1.3g/ml的浸膏;
8.s2、对浸膏进行取样并进行指纹图谱检测,色谱条件为:
9.alltech c18,4.6
×
250mm,5μm;
10.波长210nm;
11.溶剂:10%甲醇;
12.流动相体系:乙腈-0.09%磷酸;
13.流动相体系梯度为:
14.时间为0时,乙腈百分比4%,0.09%磷酸百分比为96%;
15.时间为25min时,乙腈百分比8%,0.09%磷酸百分比为92%;
16.时间为45min时,乙腈百分比9%,0.09%磷酸百分比为91%;
17.时间为55min时,乙腈百分比15%,0.09%磷酸百分比为85%;
18.时间为68min时,乙腈百分比20%,0.09%磷酸百分比为80%;
19.时间为78min时,乙腈百分比30%,0.09%磷酸百分比为70%;
20.时间为85min时,乙腈百分比35%,0.09%磷酸百分比为65%;
21.时间为90min时,乙腈百分比50%,0.09%磷酸百分比为50%;
22.时间为95min时,乙腈百分比4%,0.09%磷酸百分比为96%;
23.时间为105min时,乙腈百分比4%,0.09%磷酸百分比为96%。
24.进一步地,色谱条件还包括:流速1.0ml/min。
25.进一步地,色谱条件还包括:柱温35℃。
26.进一步地,色谱条件还包括:进样量10ul。
27.进一步地,步骤s1中,麻黄汤煎液的制备过程为:
28.称取麻黄,加水浸泡至少30min;
29.称取桂枝、蜜炙甘草和燀苦杏仁,将桂枝、蜜炙甘草和燀苦杏仁混合后加水浸泡至少30min;
30.对桂枝、蜜炙甘草和燀苦杏仁进行过滤,将滤液加入浸泡麻黄的容器中,然后大火煮沸,再调至小火煎煮10至15min;
31.将浸泡后的桂枝、蜜炙甘草和燀苦杏仁加入煎煮麻黄的容器中,共同煎煮10min后过滤,得到一次药渣和一次药液;
32.向一次药渣中加水再次煎煮20至25min,过滤得到二次药渣和二次药液,将一次药液和二次药液混合,得到麻黄汤煎液。
33.进一步地,利用不锈钢锅浸泡和煎煮麻黄。
34.本发明的有益效果是:本发明进一步优化了麻黄汤指纹图谱检测的色谱条件,并且通过试验证明了该色谱条件的准确性和有效性,在批量化生产麻黄汤的过程中,可以直接按照该色谱条件进行检测,保证检测的高效、准确。
附图说明
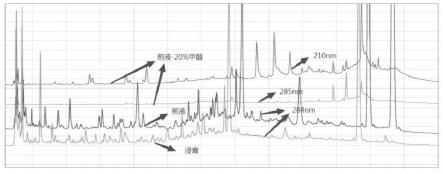
35.图1是煎液、浸膏水溶液和煎液-20%甲醇的检测结果示意图;
36.图2是浸膏在各个波长下的检测结果图;
37.图3是麻黄、苦杏仁、桂枝对照品在对应波长下的试验结果图;
38.图4是甘草混合对照品对应波长下的试验结果图;
39.图5是麻黄、苦杏仁、桂枝对照品对应波长下的试验图;
40.图6是麻黄汤煎液和浸膏在波长262nm下的对比图;
41.图7是麻黄汤煎液和浸膏在波长258nm下的对比图;
42.图8是各溶剂筛选对比图;
43.图9是甲醇/乙腈-0.09%磷酸对比图;
44.图10是乙腈-各种酸对比图;
45.图11是各波长下的指纹图谱;
46.图12是指纹图谱梯度对比图;
47.图13是指纹图谱样品和对照品位置图。
具体实施方式
48.下面结合附图和实施例对本发明进一步说明。
49.本发明的麻黄汤指纹图谱检测方法,包括
50.s1、制备麻黄汤煎液,将煎液浓缩至密度为1.2至1.3g/ml的浸膏。
51.麻黄汤煎液的制备过程为:
52.称取麻黄,加水浸泡至少30min;
53.称取桂枝、蜜炙甘草和燀苦杏仁,将桂枝、蜜炙甘草和燀苦杏仁混合后加水浸泡至少30min;
54.对桂枝、蜜炙甘草和燀苦杏仁进行过滤,将滤液加入浸泡麻黄的容器中,然后大火煮沸,再调至小火煎煮10至15min;
55.将浸泡后的桂枝、蜜炙甘草和燀苦杏仁加入煎煮麻黄的容器中,共同煎煮10min后过滤,得到一次药渣和一次药液;
56.向一次药渣中加水再次煎煮20至25min,过滤得到二次药渣和二次药液,将一次药液和二次药液混合,得到麻黄汤煎液。
57.可采用砂锅等浸泡和煎煮麻黄,优选的,本发明利用不锈钢锅浸泡和煎煮麻黄。不锈钢锅耐腐蚀,不与药材反应,并且市面上大部分的大型煎煮锅均为不锈钢锅,直接购买即可进行生产。
58.s2、对浸膏进行取样并进行指纹图谱检测,色谱条件为:
59.alltech c18,4.6
×
250mm,5μm;
60.波长210nm;
61.溶剂:10%甲醇;
62.流速1.0ml/min;柱温35℃;
63.进样量10ul;
64.流动相体系:乙腈-0.09%磷酸;
65.流动相体系梯度为:
66.时间为0时,乙腈百分比4%,0.09%磷酸百分比为96%;
67.时间为25min时,乙腈百分比8%,0.09%磷酸百分比为92%;
68.时间为45min时,乙腈百分比9%,0.09%磷酸百分比为91%;
69.时间为55min时,乙腈百分比15%,0.09%磷酸百分比为85%;
70.时间为68min时,乙腈百分比20%,0.09%磷酸百分比为80%;
71.时间为78min时,乙腈百分比30%,0.09%磷酸百分比为70%;
72.时间为85min时,乙腈百分比35%,0.09%磷酸百分比为65%;
73.时间为90min时,乙腈百分比50%,0.09%磷酸百分比为50%;
74.时间为95min时,乙腈百分比4%,0.09%磷酸百分比为96%;
75.时间为105min时,乙腈百分比4%,0.09%磷酸百分比为96%。
76.本发明中,涉及到溶液的浓度均为体积浓度,如10%甲醇、0.09%磷酸等均是以体积浓度计,乙腈百分比4%是指体积浓度为4%。
77.在指纹图谱检测中,任意一种检测条件都可能影响检测结果,因此,需要对多种检测条件进行试验,以确定较佳的检测条件。
78.实施例一
79.称取45g麻黄于不锈钢锅中,加入约300ml水浸泡45min,再称取30g桂枝、15g蜜炙甘草和30g燀苦杏仁于另一容器中加入500ml水浸泡45min,过滤,将滤液加入到浸泡麻黄的不锈钢锅中,大火煮沸,再调至小火煎煮15min,去上沫。然后再向锅中加入另3味已浸泡过的药材,共同煎煮10min,300目纱布趁热过滤,得到煎煮液和药渣。
80.向药渣中加入800ml水,大火煮沸后,再用小火(1000w)微沸25min,300目纱布趁热过滤,得到第二次煎煮液。
81.将2次煎煮液混合,得到煎液,将煎液继续加热浓缩,直至浓缩液密度为1.3g/ml,停止浓缩,得到批1浸膏。
82.实施例二
83.称取180g麻黄于不锈钢锅中,加入适量水浸泡30min,再称取120g桂枝、60g蜜炙甘草和120g燀苦杏仁于另一容器中加入适量水浸泡30min,过滤,将滤液加入到浸泡麻黄的不锈钢锅中,总体积为3200ml,大火煮沸,再调至小火(1000w)煎煮15min,去上沫。然后再向锅中加入另3味已浸泡过的药材,共同煎煮10min,300目纱布趁热过滤,得到煎煮液和药渣。
84.向药渣中加入3200ml水,大火煮沸后,再用小火微沸25min,300目纱布趁热过滤,得到第二次煎煮液。
85.将2次煎煮液混合总共6000ml(取50ml,计算干膏率),得到煎液,将煎液继续加热浓缩,直至浓缩液密度为1.3g/ml,停止浓缩,得到批2浸膏。
86.实施例三
87.称取45g麻黄于不锈钢锅中,加入约300ml水浸泡45min,再称取30g桂枝、15g蜜炙甘草和30g燀苦杏仁于另一容器中加入500ml水浸泡45min,过滤,将滤液加入到浸泡麻黄的不锈钢锅中,大火煮沸,再调至小火煎煮11min,去上沫。然后再向锅中加入另3味已浸泡过的药材,共同煎煮10min,300目纱布趁热过滤,得到煎煮液和药渣。
88.向药渣中加入800ml水,大火煮沸后,再用小火微沸21min,300目纱布趁热过滤,得到第二次煎煮液。
89.将2次煎煮液混合总体积为625ml,得到煎液,将煎液继续加热浓缩,直至浓缩液密度为1.2g/ml,停止浓缩,得到批3浸膏。
90.实施例四
91.称取180g麻黄于不锈钢锅中,加入1400ml水浸泡30min,再称取120g桂枝、60蜜炙甘草和120g燀苦杏仁于另一容器中加入600ml水浸泡30min,过滤,将滤液加入到浸泡麻黄的不锈钢锅中,水总体积为3200ml,大火煮沸,再调至小火煎煮11min,去上沫。然后再向锅中加入另3味已浸泡过的药材,共同煎煮10min,300目纱布趁热过滤,得到煎煮液和药渣。
92.向药渣中加入3200ml水,大火煮沸后,再用小火微沸21min,300目纱布趁热过滤,得到第二次煎煮液。
93.将2次煎煮液混合总共4500ml(取50ml,计算干膏率),得到煎液,将煎液继续加热浓缩,直至浓缩液密度为1.2g/ml,停止浓缩,得到批4浸膏。
94.实施例五
95.称取267.9g麻黄于不锈钢锅中,加入2600ml浸泡30min,再称取180.4g桂枝、90.4g蜜炙甘草和180.8g燀苦杏仁于另一容器中加入适量水浸泡30min,过滤,将滤液加入到浸泡麻黄的不锈钢锅中,水总体积为3600ml,大火(270℃)煮沸,再调至小火(160℃)煎煮15min,去上沫。然后再向锅中加入另3味已浸泡过的药材,共同煎煮10min,300目纱布趁热过滤,得到煎煮液和药渣。
96.向药渣中加入9300ml水,大火(270℃)煮沸后,再用小火(160℃)微沸25min,300目纱布趁热过滤,得到第二次煎煮液8600ml。
97.将2次煎煮液混合总共9800ml(大概。取50ml,计算干膏率),得到煎液,将煎液继续加热浓缩,直至浓缩液密度为1.2g/ml,停止浓缩,得到批5浸膏。
98.指纹图谱检测条件的确定:
99.1、色谱柱的考察
100.供试品溶液的制备:精密称取实施例一制备过程中的麻黄汤煎液0.1149g于25ml容量瓶中,加入10%甲醇溶剂稀释并定容至刻度,即得。
101.色谱条件:
102.sepax polar-phenyl(4.6
×
250mm,5μm)和alltech c18(4.6
×
250mm,5μm),
103.流速:1.0ml/min,
104.柱温:35℃,
105.进样量:10μl,
106.检测波长:210、285、290、207、237、254nm,
107.流动相:乙腈-0.15%磷酸,
108.按梯度2进行梯度洗脱:
109.梯度2流动相梯度表
[0110][0111]
经过试验,sepax polar-phenyl运行和alltech c18相比,在使用alltech c18色谱柱时,不同波长下得到的样品峰数量更多且峰型更好。且从全处方以及指纹图谱目的考量,需表征处方中各个药材特性,因此选择alltech c18色谱柱进行后续实验。
[0112]
从煎液样品的色谱图结果来看,各个波长下的色谱峰整体情况峰高偏低,应该与煎液样品浓度低有关;因此直接用煎液过滤进样对比煎液-20%甲醇溶剂、浸膏水溶液的色谱峰高度和峰个数来判定的结果进行后续研究。
[0113]
煎液溶剂、煎液、浸膏水溶液对比试验:
[0114]
色谱条件:
[0115]
alltech c18(4.6
×
250mm,5μm),
[0116]
流速:1.0ml/min,
[0117]
柱温:35℃,
[0118]
进样量:10μl,
[0119]
检测波长:210、285、290、207、237、254nm,
[0120]
流动相:乙腈-0.15%磷酸,
[0121]
按梯度9进行梯度洗脱。
[0122]
梯度9流动相梯度表
[0123][0124]
供试浸膏水溶液制备:精密称取批1浸膏0.2026g于10ml容量瓶中,加水稀释并定容至刻度混匀,即得。
[0125]
使用实施例一制得的煎液直接作为煎液供试样品。
[0126]
供试煎液溶液制备:精密称实施例一制得的煎液0.5536g于25ml容量瓶中,加入20%甲醇溶剂稀释并定容至刻度,即得。
[0127]
对三种供试样品进行检测,结果如图1所示。根据图1中色谱峰对比结果来看,麻黄汤浸膏色谱峰峰高与峰面积均明显大于麻黄汤煎液,因此后续实验以浸膏来开展指纹图谱研究。
[0128]
2、波长的选择
[0129]
供试品溶液制备:精密称取批1浸膏0.2026g于10ml容量瓶中,加水稀释并定容至刻度混匀,即得。煎液直接滤过进样。
[0130]
取实施例一的煎煮直接作为煎液样品。
[0131]
对照品溶液制备:
[0132]
麻黄混合生物碱对照品溶液制备:精密称取盐酸麻黄碱11.37mg、盐酸伪麻黄碱10.47mg于25ml容量瓶中,加甲醇稀释并定容至刻度,再量取1ml于10ml量瓶中,加甲醇稀释并定容至刻度,混匀,即得。盐酸麻黄碱浓度为45.48μg/ml、盐酸伪麻黄碱浓度为41.80μg/ml。
[0133]
苦杏仁苷对照品溶液制备:称取苦杏仁苷12.50mg于25ml量瓶中,加甲醇稀释并定容至刻度。混匀,即得。再精密量取1ml于10ml容量瓶中,加甲醇稀释并定容至刻度。混匀,即得。
[0134]
肉桂酸对照品溶液制备:精密称取肉桂酸9.94mg于10ml容量瓶中,加甲醇稀释并定容至刻度。混匀,即得。再精密量取1ml于10ml容量瓶中,加甲醇稀释并定容至刻度。混匀,即得。
[0135]
麻黄混合生物碱、苦杏仁苷、肉桂酸混合对照品溶液配制:精密量取麻黄混合生物碱对照品溶液、苦杏仁苷对照品溶液、肉桂酸对照品溶液各1ml于10ml容量瓶中,加甲醇定容至刻度,混匀,即得。
[0136]
甘草混合对照品溶液配制:精密称取甘草苷5.09mg,甘草酸铵5.95mg于5ml容量瓶中,加甲醇稀释溶解并定容至刻度,混匀,即得。
[0137]
色谱条件:
[0138]
alltech c18色谱柱(4.6
×
250mm,5μm),
[0139]
流速:1.0ml/min,
[0140]
柱温:35℃,
[0141]
进样量:10μl,
[0142]
流动相:乙腈-0.15%磷酸,
[0143]
检测波长根据190nm-400nm波长扫描提取的波长来选取:204、206、214、216、222、237、244、256、258、262、272、274、276、282、284nm。
[0144]
按梯度9、13、14、15进行梯度洗脱。梯度方法一致,梯度13、14、15方法的波长不一致;梯度13波长有204、206、214、216、222、228、237、244nm,梯度14波长有256、258、262、272、274、276、282、284nm,梯度15波长有258、262nm。
[0145]
梯度13流动相梯度表
[0146][0147]
梯度14流动相梯度表
[0148][0149][0150]
梯度15流动相梯度表
[0151][0152]
经过试验,浸膏在各个波长下的结果如图2所示,麻黄、苦杏仁、桂枝对照品在对应波长下的试验结果如图3所示,甘草混合对照品对应波长下的试验结果如图4所示,麻黄、苦杏仁、桂枝对照品对应波长下的试验结果如图5所示,麻黄汤煎液和浸膏在波长262nm下的对比如图6所示,麻黄汤煎液和浸膏在波长258nm下的对比如图7所示。
[0153]
根据上述色谱图可以看出,波长262nm时,各对照品溶液色谱峰个数、峰高以及处方完整性等均处于较为理想的状态,因此综合麻黄汤浸膏、煎液、各对照品溶液色谱峰个数、峰高以及处方完整性、指纹图谱表征等来看,暂时选择波长262nm进行后续指纹图谱研究。
[0154]
3、流动相体系的选择
[0155]
以麻黄混合生物碱成分的角度选取流动相体系以及磷酸浓度。
[0156]
3.1乙腈-磷酸体系中磷酸不同浓度的试验
[0157]
麻黄混合生物碱对照品溶液的制备:
[0158]
精密称取盐酸麻黄碱、盐酸伪麻黄碱各10.47mg、11.37mg于25ml容量瓶中,加甲醇稀释定容至刻度;再取1ml于10ml容量瓶中,加甲醇稀释定容至刻度即得。
[0159]
0.04%磷酸溶液:精密量取磷酸0.2ml于500ml量筒中,加水至刻度后转移至烧杯中混匀超声过滤即得。
[0160]
0.07%磷酸溶液:精密量取磷酸0.35ml于500ml量筒中,加水至刻度后转移至烧杯中混匀超声过滤即得。
[0161]
0.09%磷酸溶液:精密量取磷酸0.9ml于1000ml量筒中,加水至刻度后转移至烧杯中混匀超声过滤即得。
[0162]
0.15%磷酸溶液:精密量取磷酸0.75ml于500ml量筒中,加水至刻度后转移至烧杯中混匀超声过滤即得。
[0163]
水:量取500ml水过滤即得。
[0164]
色谱条件:
[0165]
alltech c18色谱柱(4.6
×
250mm,5μm),
[0166]
流速:1.0ml/min,
[0167]
柱温:35℃,
[0168]
进样量:10μl,
[0169]
检测波长:262nm,
[0170]
流动相:乙腈-磷酸(0.04%、0.07%、0.09%、0.15%)、乙腈-水。
[0171]
按梯度4进行梯度洗脱。
[0172]
梯度4流动相梯度表
[0173][0174]
从麻黄混合生物碱成分色谱图结果来看,磷酸溶液浓度为0.09%时峰型相对于其他浓度较好;因此水相中选取0.09%浓度定为磷酸浓度。
[0175]
3.2流动相体系中有机相、水相中酸的种类选择
[0176]
麻黄混合生物碱对照品溶液配制:精密称取盐酸麻黄碱、盐酸伪麻黄碱各13.94mg、10.45mg于10ml容量瓶中,加甲醇稀释定容至刻度即得。
[0177]
色谱条件:
[0178]
dikma c18(4.6
×
250mm,5μm)色谱柱,
[0179]
流速:1.0ml/min,
[0180]
柱温:35℃,
[0181]
进样量:10μl,
[0182]
检测波长:262nm。
[0183]
流动相体系1:以甲醇/乙腈-0.09%磷酸为流动相,流动相梯度同梯度4。
[0184]
流动相体系2:以乙腈-0.09%甲酸为流动相,流动相梯度同梯度4。
[0185]
流动相体系3:以乙腈-0.09%冰乙酸酸为流动相,流动相梯度同梯度4。
[0186]
经过试验,甲醇/乙腈-0.09%磷酸对比如图9所示,乙腈-各种酸对比如图10所示,从对比图来看,甲醇和乙腈对比,乙腈的洗脱要比甲醇强一些;因此选乙腈为有机相;乙腈和各种酸对比图来看,甲酸和冰乙酸相比磷酸在基线、峰型上都要差,因此选磷酸确定为酸的种类;结合上述磷酸浓度的确定,因此确定乙腈-0.09%磷酸体系作为确定的流动相体系。
[0187]
4、溶剂的选择
[0188]
供试品的制备:精密称取批3浸膏样品1.5226g、1.2938g、1.1312g、1.1766g、1.1271g溶于各10%甲醇、20%甲醇、30%甲醇、40%甲醇、50%甲醇溶液中,并定容于相应于25ml容量瓶中,加相应溶剂稀释溶解并定容于刻度混匀得到供试品溶液。
[0189]
色谱条件:
[0190]
dikma c18(4.6
×
250mm,5μm)色谱柱,
[0191]
流速:1.0ml/min,
[0192]
柱温:35℃,
[0193]
进样量:10μl,
[0194]
波长:262nm,
[0195]
流动相体系:乙腈-0.09%磷酸。
[0196]
流动相梯度同梯度4。
[0197]
各溶剂筛选对比如图8所示,从图中可以看出,采用10%甲醇时,各峰的完整性最高,因此选择10%甲醇定为指纹图谱样品的提取溶剂。
[0198]
5、梯度优化
[0199]
供试品的制备:精密称取批4浸膏样品1.5982g于25ml容量瓶中,加10%甲醇溶剂稀释溶解并定容于刻度,混匀得到供试品溶液。
[0200]
麻黄汤指纹图谱混合对照品溶液配制:精密称取盐酸麻黄碱11.10mg、盐酸伪麻黄碱13.13mg、苦杏仁苷9.34mg、肉桂酸9.94mg、甘草苷7.41mg、甘草酸铵8.35于各10ml容量瓶中,加甲醇稀释定容至刻度,混匀即得;再精密量取各对照品溶液1ml于25ml容量瓶中,加甲醇稀释定容至刻度,混匀即得。
[0201]
色谱条件:
[0202]
dikma c18(4.6
×
250mm,5μm)色谱柱,
[0203]
流速:1.0ml/min,
[0204]
柱温:35℃,
[0205]
进样量:10μl,
[0206]
检测波长:262、210、220、230nm,
[0207]
进行梯度13、15、30、31、34优化。
[0208]
各波长下的指纹图谱如图11所示,在进行梯度优化之前对比了前面各波长关于麻黄混合生物碱、苦杏仁苷的峰高,发现其峰高太低是因为不同波长下吸收不一样,虽能够体现,但不是很明显;且麻黄、苦杏仁药材在处方中为君臣关系,根据其功效和指纹图谱原则还是将其定为主要的成分体现在指纹图谱上;因此重新运行波长210、220、230nm对比波长262nm,麻黄和苦杏仁成分还是集中在低波长中,因此选择吸收较为高的波长210nm进行后续梯度优化。
[0209]
梯度13、15见上文梯度13表和梯度15表,梯度30、31、34如下所示:
[0210]
梯度30流动相梯度表
[0211][0212]
梯度31流动相梯度表
[0213][0214][0215]
梯度34流动相梯度表
[0216][0217]
经试验,指纹图谱梯度对比如图12所示,指纹图谱样品和对照品位置如图13所示。从图中确定了梯度34作为指纹图谱梯度,各对照品成分位置得到对应,基线平稳,麻黄混合生物碱分离度为1.7,苦杏仁苷分离度为1.3,甘草苷分离度为2.1,肉桂酸分离度为6.6,甘草酸分离度为5.6。
[0218]
综上,麻黄汤浸膏的色谱条件为:
[0219]
alltech c18,4.6
×
250mm,5μm;
[0220]
波长210nm;
[0221]
溶剂:10%甲醇;
[0222]
流速1.0ml/min;柱温35℃;
[0223]
进样量10ul;
[0224]
流动相体系:乙腈-0.09%磷酸;
[0225]
流动相体系梯度为:
[0226][0227]
本发明通过多次试验,确定了合理的波长、溶剂、流动相体系以及流动相体系梯度,在批量化生产过程中,可以按照本发明的色谱条件对麻黄汤浸膏进行取样检测,可以准确检测产品的药效等是否满足要求,从而判断产品是否合格,从而保证产品药效。
[0228]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
