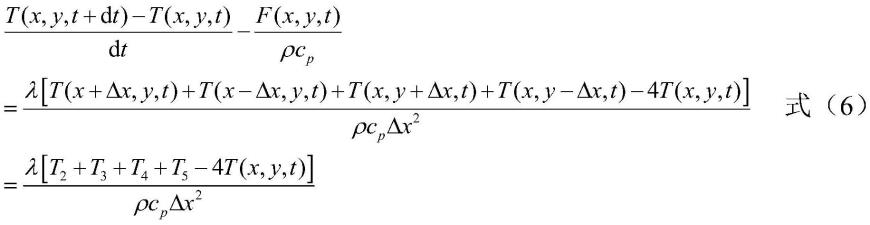
1.本发明涉及界面作用研究技术领域,尤其涉及一种基于离散粒子-有限元网格耦合模型的界面计算方法及装置。
背景技术:
2.高超声速飞行器在临近空间滑翔、飞行或再入过程中,由于强烈的激波和黏性摩擦阻力而产生高温气体效应,导致激波层内气体分子的内能被激发,粒子发生解离、电离等反应,进一步引发飞行器热防护系统近壁面发生复杂的物理化学作用过程,对飞行器气动力特性和气动热环境造成严重影响,给飞行器表面材料的防热设计带来极大挑战。
3.气固多相催化反应或液固多相催化反应中,气体在催化剂材料内部和表面发生反应,产生复杂的吸热/放热和传热等过程,导致催化剂材料温度发生变化,严重影响了催化剂的性能,为反应材料的研制和反应过程的放大设计带来挑战。
4.因此,气固表面相互作用或液固表面相互作用是航空航天和材料化学领域研究的重要问题,它在多相催化、吸附、分离、高超声速飞行器热防护系统、稀薄气体动力学等很多领域具有广泛的应用前景。目前,对气固表面上发生的复杂物理化学过程中产生的能量转移和转化过程进行建模和研究存在着很大的困难,现有模型精确性差,而且难以对表面上发生的各微观过程进行深入研究。
5.目前对气固表面相互作用主要通过下面几种模型和方法进行处理。一种是将界面一侧(或两侧)的物质用基于连续介质假设的模型和方法进行描述,比如用计算流体力学方法描述气体,然后将两者的相互作用处理为一种边界条件,比如将固体边界看作滑移/无滑移边界条件或者理想粘性壁面等,并用某些给定的假设参数进行模拟。
6.cn106055756a公开了一种速度滑移效应下静压气浮导轨系统仿真分析方法,将速度滑移计算方法及边界条件引入雷诺方程,推导出来适合微尺度下压缩气体的雷诺方程;通过cfx仿真分析能够得到不同节流孔直径对气膜厚度的影响,不同气膜厚度下,气浮导轨压强的变化,通过与传统雷诺方程仿真出的结果作对比,能够更准确的确定导轨的静特性,此方法简单效率高,更加有利于指导静压导轨应用于实践。
7.在高超声速飞行器气固表面作用的研究中,也有考虑基于有限速率反应的模型对固体表面进行描述,此时的固体壁面被看作是带特定反应参数的边界条件,并用反应系数、催化效率等参数来描述气固表面发生化学反应产生的热效应。这些方法在描述气固相互作用尤其是能量转移转化过程时,给定的参数依赖于经验/半经验模型或者有限的实验结果,不具有普适性,难以扩展到不同气体、固体材料或者不同模拟条件的情况,对新的体系的预测偏差和不确定性都很大。
8.最重要的是,上述的模型和方法都没有考虑气固表面相互作用过程中化学反应与传热相互耦合的过程,因此不能准确获得不同形式能量间的转移和转化过程,模型对热流和温度分布的预测也会出现较大的偏差。
9.因此,需要开发一种新的界面计算方法,提高计算结果的准确性。
技术实现要素:
10.鉴于现有技术中存在的问题,本发明提供一种基于离散粒子-有限元网格耦合模型的界面计算方法及装置,所述方法能够对气固表面作用过程中包括物理过程、化学反应和传热在内的多种过程间的耦合以及不同形式能量和物质之间的转移和转化过程进行建模和模拟计算,所述方法可以用于飞行器表面防热材料研制、气动热精确预测或催化剂反应动力学等多个领域的研究。
11.为达此目的,本发明采用以下技术方案:
12.第一方面,本发明提供一种基于离散粒子-有限元网格耦合模型的界面计算方法,所述界面计算方法包括如下步骤:
13.(1)针对界面体系,对界面一侧的气相和/或液相建立离散粒子模型,对界面另一侧的固相建立有限元网格模型;
14.(2)根据气相和/或液相与固相之间实际的作用过程,选取界面之间传质、传热或反应中任意一种或至少两种组合的作用过程,并对所述作用过程选择特定的模型方程;
15.(3)根据气相和/或液相与固相之间物质和/或能量的转变过程,以步骤(3)中所述模型方程为基础建立物质和/或能量在离散粒子模型和有限元网格模型之间的关联,完成计算模型的构建;
16.(4)根据步骤(3)所述计算模型进行模拟计算。
17.本发明提供的界面计算方法根据界面两边物质的种类和特性分别采用离散粒子和有限元网格进行描述,然后根据粒子和表面相互作用中的扩散、吸附、脱附、反应机理、反应条件以及传热过程等选择模型方程;在模型中对以上过程中不同形式的能量以及相互之间的转移和转化过程用参数进行描述;建立这些参数在粒子和网格之间的关联,从而构建界面处能量和物质的转移和转化过程的耦合计算模型,建立的计算模型考虑了不同形式能量间的关联,从而实现了表面相互作用过程中能够包括化学反应与传热等在内的多过程耦合模拟。
18.本发明提供的界面计算方法相较于传统的连续介质假设的模型和方法,其中界面相互作用的参数和边界条件无需经验或半经验的实验结果支撑,具有更佳的普适性,且能够拓展到不同气体、固体或液体,或者拓展到不同的模拟条件,而且能够同时考虑反应、传热和传质三种耦合过程,模拟计算更精确。相较于微观分子动力学模拟而言,本发明的计算量显著降低,且能够适用于较大的时空尺度,不仅能够得到宏观的浓度分布场和温度分布场数据,而且能够得到动力学演化的过程,且相较于传统的md方法,能够适用于反应计算过程。
19.优选地,步骤(1)中所述离散粒子模型中离散粒子包括质点粒子、规则几何结构粒子或不规则几何结构粒子中的任意一种或至少两种的组合,其中典型但非限制性的组合为质点粒子和规则几何结构粒子的组合,规则几何结构粒子和不规则几何结构粒子的组合,质点粒子和不规则几何结构粒子的组合。
20.优选地,所述离散粒子中每一个粒子表示一个原子、一个分子、原子团簇或分子团簇。
21.优选地,步骤(1)中所述离散粒子的参数包括质量、位置、原子直径、分子直径、平动速度、热速度、化学键键能、悬挂键键能、吸附热、转动能、振动能或电子能中的任意一种或至少两种的组合,其中典型但非限制性的组合为质量和位置的组合,原子直径和平动速度的组合,原子直径和热速度的组合,平动速度和化学键键能的组合,热速度和悬挂键键能的组合,化学键键能和吸附热的组合,吸附热和转动能的组合。本发明中优选提供越多的参数,模拟计算的结果越准确,但当某些参数缺少时也可根据其他参数进行模拟计算。
22.优选地,步骤(1)中所述有限元网格模型包括二维表面网格模型或三维网格模型。
23.优选地,所述固相的原始结构包括规则几何表面、不规则几何表面、实体结构、多孔材料结构或者粒子堆积结构中的任意一种或至少两种的组合,其中典型但非限制性的组合为规则几何表面和不规则几何表面的组合,不规则几何表面和实体结构的组合,不规则几何表面和粒子堆积结构的组合,实体结构和粒子堆积结构的组合,粒子堆积结构和多孔材料结构的组合。
24.优选地,步骤(1)中所述有限元网格模型的参数包括网格形状、网格尺寸、材料密度、材料比热容、材料热导率、材料表面辐射系数、网格吸收热量、网格放出热量和材料温度。
25.优选地,所述有限元网格模型的参数还包括反应位点的尺寸和反应位点的形状。
26.优选地,所述有限元网格模型的参数还包括吸附位点的尺寸和吸附位点的形状。
27.本发明中所述反应位点的尺寸和形状包括对空间中点、线、面、体的描述和数据坐标。
28.优选地,步骤(2)所述传质包括扩散、吸附或脱附中的任意一种或至少两种的组合,其中典型但非限制性的组合为扩散和吸附的组合,扩散和脱附的组合,吸附和脱附的组合,扩散、吸附和脱附三者的组合。
29.优选地,所述传热包括热辐射、热传导或热对流中的任意一种或至少两种的组合,其中典型但非限制性的组合为热辐射和热传导的组合,热辐射和热对流的组合,热传导和热对流的组合,热辐射、热对流和热传导三者的组合。
30.优选地,所述反应包括气相和/或液相反应、气固催化反应、液固催化反应或气液固三相催化反应中任意一种或至少两种的组合,例如可以是气液反应和气固催化反应的组合,气固催化反应和液固催化反应的组合,气气反应或液液反应的组合。
31.本发明中离散粒子之间发生的相互作用过程不做特别指定,可以按照特定的物理和化学机理模型对物质和能量的转移和转化过程进行建模处理。同样,固体材料网格内的能量转移和转化过程也不做特别指定,可以按照给定的传热过程模型进行处理。
32.优选地,步骤(3)所述计算模型采用真实时空尺度或无量纲的尺度。
33.优选地,所述能量包括动能、势能、化学能、电磁能、辐射能或热能中的任意一种或至少两种的组合,其中典型但非限制性的组合为动能和势能的组合,动能和辐射能的组合,势能和化学能的组合,化学能和电磁能的组合,电磁能和热能的组合。
34.优选地,步骤(3)中能量在离散粒子模型和有限元网格模型之间的关联包括:在每一个时间步长内每一个网格的能量变化包括第一能量变化和第二能量变化。所述第一能量变化为所述时间步长内离散粒子对所述网格的第一能量作用;所述第二能量变化为相邻网格对所述网格的第二能量作用。
35.本发明的离散粒子与网格发生相互作用时,粒子与网格间的能量交换影响网格的温度变化,而网格温度的变化又影响粒子在网格上的扩散、吸附、反应或脱附过程,因此两个过程相互耦合。
36.根据物理和化学反应的机理和过程,能量将在离散粒子和网格间进行传递。离散粒子的能量主要包括动能、势能、化学键键能以及内能(这里的内能指颗粒内部的转动能、振动能和电子能);粒子与网格发生作用时,除了碰撞作用外,还要根据描述网格是否具有吸脱附位点和反应位点判断当前网格是否发生吸脱附和反应过程。相互作用的过程包括但不限于碰撞、吸附、化学反应或脱附等过程。每个网格可以包含一个或多个作用位点,所述作用位点包括吸脱附位点和/或反应位点,在每一次作用过程中,粒子与网格反应位点之间交换的能量(比如化学反应吸/放热、碰撞交换能量等)可以分别记为dqi,其正负可以自行约定,比如设定位点吸热为正,位点放热为负。在上述作用的过程中,位点的温度会影响dqi的大小,而位点传递给粒子的能量立即作用或标记在粒子对应的参数上。在每一个时间步长内,网格与网格之间的热传导以及网格热辐射的能量记为dqj,其正负约定同上。
37.因此,在一个时间步长内,每个网格所有位点的吸/放热量之和记为dq,且满足dq=∑dqi ∑dqj,因此每一个网格都可以看作是一个热源,从而可以和固体材料自身的传热模型相耦合。
38.优选地,所述第一能量作用包括碰撞交换能量、反应吸热、反应放热、吸附热、脱附热或热对流中的任意一种或至少两种的组合,其中典型但非限制性的组合为碰撞交换能量和反应吸热的组合,反应吸热和吸附热的组合,吸附热和脱附热的组合,热对流和碰撞交换能量的组合,碰撞交换能量和脱附热的组合。
39.优选地,所述第二能量作用包括热传导和/或热辐射。
40.优选地,步骤(3)中物质在离散粒子模型和有限元网格模型之间的关联包括:在每一个时间步长内每一个网格的物质变化包括第一物质变化;所述第一物质变化包括吸附和/或脱附。
41.优选地,所述第一物质变化还包括反应的物质交换。
42.优选地,步骤(3)中还包括初始条件和边界条件的建立。
43.根据具体的物理过程机理或化学反应机理或者简化的物理或化学反应过程,建立包含空间区域、材料结构、物理过程和/或化学过程、初始条件和边界条件等在内的计算体系模型及确定相应的模拟参数。具体包括离散粒子参数和有限元网格参数以及温度、压力、初始条件、边界条件,吸附类型、化学反应路径、化学反应网络、能量转移或转化路径和大小等,从而生成计算模型。
44.优选地,针对稳定来源气相和/或液相,在计算模型中设置组分控制区域。
45.本发明针对稳定来源的气相和/或液相,通过设置组分控制区域,能够保障区分控制区域的气相和/或液相的温度和组分不发生变化,而又能够与模拟区域的组分进行实时的扩散和交换,适用于模拟流体与飞机表面的相互作用等情况。
46.本发明对所述模拟计算的方法和平台没有特殊限制,可采用本领域技术人员熟知的方法和平台进行计算,也可根据实际情况自行设计程序进行模拟计算。
47.第二方面,本发明提供基于离散粒子-有限元网格耦合模型的界面计算装置,所述界面计算装置能够实现第一方面所述的基于离散粒子-有限元网格耦合模型的界面计算方
法。
48.本发明提供的界面计算装置能够以较短的算力实现较高准确度的模拟计算,不仅能够实现反应、传质和传热过程的耦合,而且能够得到动力学演化过程,应用前景广阔。
49.优选地,所述界面计算装置包括离散粒子模型建立模块、有限元网格模型建立模块、模型方程选取模块、计算模型构建模块和模拟计算模块。
50.所述离散粒子模型建立模块、有限元网格模型建立模块、模型方程选取模块分别得到的离散粒子模型、有限元网格模型和模型方程用于计算模型构建模块构建计算模型;所述模拟计算模块采用计算模型进行计算。
51.与现有技术相比,本发明至少具有以下有益效果:
52.(1)本发明提供的基于离散粒子-有限元网格耦合模型的界面计算方法能够适用于大时空尺度的模拟计算且算力时间显著缩短;
53.(2)本发明提供的基于离散粒子-有限元网格耦合模型的界面计算方法能够得到各个过程,如吸附、反应、脱附或辐射等中能量交换随时间的变化、离散粒子参与不同过程的情况以及对其传热的贡献情况等,可适用于不同物系,普适性高,而且无需依赖于经验/半经验模型或者有限的实验结果,而且可以对各类复杂的气相和/或液相与固相作用过程中物质和能量的转移和转化过程进行建模和模拟;同时,根据实际运行条件等的改变,还能够方便的对气体氛围、材料属性以及气固作用条件等进行修改,更新运行结果;
54.(3)本发明提供的基于离散粒子-有限元网格耦合模型的界面计算装置能够以较短的算力实现较高准确度的模拟计算,能够实现反应、传质和传热过程的耦合,可以用于飞行器表面防热材料研制、气动热精确预测以及催化剂反应动力学等多个领域的研究,应用前景广阔。
附图说明
55.图1是实施例1建立的离散粒子模型与有限元网格模型的组合示意图。
56.图2是实施例1中采用五中心点有限差分方式的示意图。
57.图3是应用1计算得到的sio2表面上的o原子的损失效率随时间的变化图。
58.图4是文献计算得到的sio2表面上的o原子的损失效率随时间的变化图。
59.图5是应用2中计算得到的固体表面温度随时间的变化图。
60.图6是应用2计算得到的吸附原子在固体的表面覆盖率随时间的变化图。
61.图7是应用2计算得到的气固作用各过程中吸/放热的热流密度随时间的变化图。
62.图8是应用3中每百步耗时随中央处理核心数的变化及拟合图。
63.图9是图8中拟合曲线拓展至100万个中央处理器核心数的变化图。
64.图中:1-二维几何平面模型;2-模拟计算区;3-组分控制区。
具体实施方式
65.下面结合附图并通过具体实施方式来进一步说明本发明的技术方案。
66.下面对本发明进一步详细说明。但下述的实例仅仅是本发明的简易例子,并不代表或限制本发明的权利保护范围,本发明的保护范围以权利要求书为准。
67.本发明例提供一种基于离散粒子-有限元网格耦合模型的界面计算方法,所述界
面计算方法包括如下步骤:
68.(1)对气相和/或液相建立离散粒子模型,对固相建立有限元网格模型;
69.(2)根据气相和/或液相与固相之间实际的作用过程,选取界面之间传质、传热或反应中任意一种或至少两种组合的作用过程,并对所述作用过程选择特定的模型方程;
70.(3)根据气相和/或液相与固相之间物质和/或能量的转变过程,以步骤(3)中所述模型方程为基础建立物质和/或能量在离散粒子模型和有限元网格模型之间的关联,完成计算模型的构建;
71.(4)根据步骤(3)所述计算模型进行模拟计算。
72.实施例1
73.本实施例提供一种基于离散粒子-有限元网格耦合模型的界面计算方法,所述界面计算方法以空气和二氧化硅材料的催化反应过程为例,具体过程描述为:高温环境下,空气发生离解,气体在二氧化硅材料表面发生复杂的催化反应过程,释放大量热量造成材料温度升高。
74.所述界面计算方法具体包括如下步骤:
75.(1)对高温空气气体建立离散粒子模型,设置的参数包括:气体密度为0.032kg/m3;离散颗粒的质量和直径分别为:o2(5.3138
×
10-23
g,0.13996nm)、n2(4.6496
×
10-23
g,0.15027nm)、no(4.9817
×
10-23
g,0.14560nm)、o(2.6569
×
10-23
g,0.12362nm)、n(2.3248
×
10-23
g,0.11956nm);气体原子在表面的吸附热分别为:o(499.8kj/mol)、n(530.8kj/mol);各气体分子的键能分别为:o2(498.36kj/mol)、n2(945.33kj/mol)、no(630.57kj/mol);
76.对二氧化硅材料建立有限元网格模型;具体地,二氧化硅材料为具有一定厚度的薄片结构,不考虑材料内沿厚度方向的温度梯度,因此采用二维几何平面模型表示,如图1所示。在二维几何平面模型1上部有模拟计算区2,x和y方向为周期性边界条件,长度均为70nm,z方向厚度为1μm,最上方厚度为20nm的区域为组分控制区3,以保证该区域内气体的温度和组分等不变,模拟稳定的来流气体;固体上的反应位点密度为4.5个/nm2。固体厚度为10nm,二维网格边长尺寸为10nm;固体表面辐射系数ε为0.8,密度ρ为2.2g/cm3,比热c
p
为1124j/(kg
·
k),导热系数λ为7.6w/(m
·
k);
77.(2)根据气相和/或液相与固相之间实际的作用过程,选取界面之间传质、传热和反应三种组合的作用过程,并对所述作用过程选择特定的模型方程;
78.其中,所述传质为碰撞、吸附和脱附三种作用过程;所述传热为热辐射和热传导;所述反应为空气气体分子离解后的原子在固体表面上发生的复合反应;
79.根据传质和反应特性,选择朗格缪尔-欣谢尔伍德(langmuir-hinshelwood(l-h))模型方程和eley-rideal(e-r)模型方程,其中e-r为自发反应,而l-h反应的能垒分别为:o-o反应(300.96kj/mol)、o-n反应(243.3kj/mol)、n-n反应(72.18kj/mol);
80.单位时间、单位面积上表面向空间辐射的热量为σεt4,其中,斯忒藩-玻耳兹曼常数σ为5.67032
×
10-8
(w/m2.k4),ε为固体表面辐射系数;热传导采用二维有源热传导方程进行描述,即如下式(1)所示:
[0081][0082]
其中,t为温度,f(x,y,t)表示不同时间和位置的热源强度,t表示时间,ρ为固体的
密度,c
p
为固体材料的比热容,λ为导热系数。
[0083]
(3)根据气相和/或液相与固相之间物质和/或能量的转变过程,以步骤(3)中所述模型方程为基础建立物质和/或能量在离散粒子模型和有限元网格模型之间的关联,完成计算模型的构建;
[0084]
具体地,所述转变过程包括原子吸附、分子吸附、化学反应、气固碰撞和热脱附,具体如下:
[0085]
原子吸附:原子在反应位点作用且满足(物理或化学)吸附条件时,原子向反应位点释放热量,吸附后原子的运动速度根据此时反应位点温度下的偏maxwell速度分布随机生成的速度来确定,其中偏maxwell速度由如下公式(2)~(4)确定,其中,u、v、w分别是颗粒在三个方向上的速度分量,k为玻尔兹曼常数,m是颗粒质量,tw为固体表面温度,ranf1~ranf5是五个相互独立的随机数,且其取值范围均为[0,1],然后总动能ek如式(5)所示。下面的模型中涉及到由表面温度确定粒子运动速度(或者动能)的均按照该方法进行:
[0086][0087][0088][0089][0090]
由于吸附后的原子只能在固体表面做二维运动,自由度减少,因此设置法向速度u=0,故原子的实际运动速度只取决于三分之二的平动能。此时再根据总能量守恒,由larsen-borgnakke模型对能量在不同自由度上进行重新分配,并计算固体反应位点吸收或者释放的热量;
[0091]
分子吸附:分子在表面可以发生物理吸附或者化学吸附,当发生化学吸附时,分子需要具备足够大的能量首先能够发生化学键断裂,且表面上需要具有两个相邻的可吸附位点供生成的原子分别发生吸附,该过程中吸收和放出的总能量类比于原子吸附过程进行计算;
[0092]
化学反应:当两个已经吸附在表面的吸附原子相遇时,会发生反应生成分子并离开表面(l-h反应过程),该过程将从表面吸收能量,大小为两个原子的吸附热与反应热之差;根据总能量守恒,计算新生成分子的能量,新生成的分子离开表面时的动能根据表面温度确定,并对其内能进行重新分配。当一个气相原子与一个已经吸附的原子相遇时,可能会发生反应生成分子并离开表面(e-r反应过程),该过程将从表面吸收能量,大小为原子吸附热与分子化学键键能之差,其余处理方式同上。
[0093]
气固碰撞:离散颗粒与表面发生作用时,如果不满足上述吸附和反应条件,则将表面作为边界进行碰撞过程处理,碰撞模型采用漫反射模型,颗粒碰撞反弹后的速度根据壁面温度确定,然后再由总能量守恒确定从壁面吸收或释放的能量。
[0094]
热脱附:当壁面温度升高后,吸附原子具有的能量足够高,大于原子的吸附热时则发生热脱附而离开表面,此时从反应位点吸收能量,大小为一个吸附热。
[0095]
能量传递:在一个时间步长内,每个网格所有位点的吸/放热量之和记为dq,且满足dq=∑dqi ∑dqj;粒子与网格反应位点之间交换的能量(比如化学反应吸/放热、碰撞交换能量等)可以分别记为dqi;网格与网格之间的热传导以及网格热辐射的能量记为dqj。
[0096]
(4)根据步骤(3)所述计算模型进行模拟计算,如图2所示,所述模拟计算中采用五中心点有限差分方式求解式(1)中的方程,其中式(1)中方程的离散形式如下式(6)所示:
[0097][0098]
式(6)中t(x,y,t)表示不同时间不同位置的温度,
△
x表示五中心点有限差分中的x方向和y方向的位移差。
[0099]
具体应用如下:
[0100]
应用1
[0101]
本应用在实施例1提供的模型和参数的基础上,采用的具体来流气体为包含o和o2的高温气体(摩尔比为1:9),固体采用等温壁面条件分别模拟了不同个温度下o原子的损失效率,进行模拟计算,采用的平台和平台配置情况如表1所示。
[0102]
表1
[0103]
平台mole-8.5e中央处理器(cpus)intel xeon e5-2680 v2(2.80ghz,10cores)操作系统(os)centos release 6.3(final)kernel:2.6.32-279.el6编译器(compiler)gcc:4.4.6,intel compiler:14.0.0
[0104]
本应用的计算结果如图3所示,从图3中能够很清楚的看出,低温下sio2表面上的o原子的损失主要是由于e-r反应贡献,在高温下,随着壁面温度升高,l-h反应越来越重要。
[0105]
为验证本发明模拟计算的有效性,将本应用的计算结果与matthew等(matthew maclean,“finite-rate surface chemistry model,i:formulation and reaction system examples,”42nd aiaa thermophysics conference,2011.)的计算结果相比较,文献的模拟结果如图4所示,从图3和图4中可以看出,本发明的结果与已有文献吻合很好,表明了本模型的有效性和准确性。
[0106]
应用2
[0107]
本应用在实施例1的模型和参数的基础上,采用的高温气体采用5组分空气,即包含氧分子(o2)、氮分子(n2)、氧原子(o)、氮原子(n)以及一氧化氮分子(no),气体的初始温度为5700k,初始固体温度为250k;气体各组分的摩尔比为:
[0108]
n:o:n2:o2:no=0.1051384:0.325525:0.5580423:0.0004246:0.0108697
[0109]
固体表面为自由对流和辐射边界条件,对气固反应过程进行模拟和计算。
[0110]
图5给出的是固体表面温度随时间的变化,从图5能得到随着反应时间的进行,固体表面温度的升温情况。
[0111]
图6给出的是吸附原子在表面的覆盖率随时间的变化,从图6可以看出,随着时间的进行,在固体表面吸附的原子在短时间内能够迅速吸附至固体表面,然后随着反应的进行,反应产生的气相从固体表面脱附,或随着固体表面温度的升温,原本吸附在固体表面的原子从固体表面脱附造成后续固体表面覆盖率的显著下降,但随着反应达到平衡,固体表面的覆盖率进一步不再发生变化。
[0112]
图7给出的是气固作用各过程中吸/放热的热流密度随时间的变化,从图7中能够直接看出在某一时间段,热量交换主要由哪一具体过程造成,为进一步优化实际反应过程或对反应进行精准预测提供了基础数据。
[0113]
由此可以看出,通过本发明的模拟,能够分析不同阶段下哪个过程对能量交换贡献最大、吸附原子参与各过程的比例以及表面催化系数等结果,从微观角度为气动热环境的精确预测提供了新的研究模型和手段。
[0114]
应用3
[0115]
本应用采用应用2的体系开展并行计算,对本发明所需的算力进行评估。本应用中单进程计算规模为70nm
×
70nm
×
1μm,单进程包含粒子数约5000个。保持z方向规模不变,扩大x和y方向的尺寸,分别测试了160、200、240、280、320、360和400个cpu核心下,体系每演化100时间步长(1ps)所耗费的物理时间,结果如图8所示,160个cpu核心数体系演化100时间步(1ps)所需的物理时间仅为0.09133秒。按照当前的并行效率,推测模拟规模达到100万个cpu核心数的百步耗时为0.10801秒,如图9所示,预测百万核心并行计算时其并行效率为81.74%,表明本模型具有较好的并行性。
[0116]
由此可以看出,本发明提供的界面计算方法尽管在模拟过程中设置有大量气体分子,仍然具有非常高效的并行效率以及短的模拟时间,远高于采用势函数的分子动力学方法的效率。
[0117]
综上所述,本发明提供的基于离散粒子-有限元网格耦合模型的界面计算方法能够对气固表面作用过程中包括化学反应和传热在内的多种过程间的耦合以及不同形式能量和物质之间的转移和转化过程进行建模和模拟计算,100万个cpu核心数的并行效率可达到81.74%以上,且预测结果准确可靠,大大缩短了模拟计算时长。
[0118]
申请人声明,本发明通过上述实施例来说明本发明的详细过程和模型算法特征,但本发明并不局限于上述详细结构特征,即不意味着本发明必须依赖上述详细模型和算法特征才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用过程模型的等效替换以及辅助模型的增加、具体计算方式或计算模型的选择等,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。