1.本发明涉及纳米压印用抗蚀剂下层膜形成用组合物、作为由该组合物形成的涂布膜的固化物的抗蚀剂下层膜、该抗蚀剂下层膜的制造方法、以及利用了该抗蚀剂下层膜的图案形成方法以及半导体装置的制造方法。
背景技术:
2.在要求微细化的半导体器件、mems等的制造中,可以在基板上形成数纳米级的微细的结构体的光纳米压印技术受到关注。这是如下的技术:在基板(晶片)上涂布固化性组合物(抗蚀剂),向其按压表面形成了微细的凹凸图案的模具(模),在该状态下直接使抗蚀剂通过热或光而固化,将模具的凹凸图案转印于抗蚀剂固化膜,将模具拉离,将图案形成在基板上。
3.对于一般的光纳米压印技术,首先,在基板上的图案形成区域使用喷墨法等滴加液态的抗蚀剂组合物,将抗蚀剂组合物的液滴在基板上扩展(预展开)。接下来,将该抗蚀剂组合物使用相对于照射光为透明并进行了图案形成的模具(模)来进行成型。此时,抗蚀剂组合物的液滴通过毛细管现象而向基板与模具的间隙的整个区域扩展(展开)。此外,抗蚀剂组合物通过毛细管现象也向模具上的凹部的内部填充(装填)。展开和装填完成为止的时间为填充时间。在抗蚀剂组合物的填充完成后,照射光而使抗蚀剂组合物固化,接着使两者分离。通过实施这些工序,从而在基板上形成具有规定的形状的抗蚀剂的图案。
4.在光纳米压印技术的脱模工序中,抗蚀剂组合物与基材之间的密合性是重要的。因为如果抗蚀剂组合物与基材之间的密合性低,则有时在脱模工序中在将模具分离时,发生使抗蚀剂组合物固化而获得的光固化物的一部分附着于模具的状态下剥落的、图案剥落缺陷。作为使抗蚀剂组合物与基材之间的密合性提高的技术,提出了在抗蚀剂组合物与基材之间形成作为用于使抗蚀剂组合物与基材密合的层的密合层的技术。
5.此外,有时纳米压印中的图案形成使用高蚀刻耐性层。作为高蚀刻耐性层的材料,一般使用有机系材料、有机硅系材料。进一步,可以在纳米压印用抗蚀剂下层膜上,通过涂布或蒸镀而形成密合层、包含si的有机硅层。在这些密合层、包含si的有机硅层为疏水性、且显示高的纯水接触角的情况下,下层膜也为疏水性、且显示高的纯水接触角的情形可以期待膜间的密合性提高,不易剥离。
6.由于已知he、h2、n2、空气等在室温下为比较疏水性,因此期待与高接触角的膜的亲和性高、气体透过性提升。因此,作为下层膜材料,也优选为水接触角高的物质。
7.现有技术文献
8.专利文献
9.专利文献1:日本特开2019-36725号公报
技术实现要素:
10.发明所要解决的课题
11.因此,本发明所要解决的课题是提供显示良好的平坦化性、通过烧成而获得具有高疏水性的膜、能够提高与疏水性的上层膜的密合性、而且可以通过变更树脂的分子骨架来调整为适应于工艺的光学常数、蚀刻速度的纳米压印用抗蚀剂下层膜形成用组合物。
12.用于解决课题的方法
13.本发明包含以下方案。
14.[1]一种纳米压印用抗蚀剂下层膜形成用组合物,其包含具有下述式(1)所示的重复单元结构的酚醛清漆树脂。
[0015][0016]
[在式(1)中,
[0017]
基a表示具有芳香族环、稠合芳香族环、或稠合芳香族杂环的有机基,
[0018]
基b表示具有芳香族环、或稠合芳香族环的有机基,
[0019]
基e表示单键、或者可以被取代且可以包含醚键和/或羰基的支链或直链的碳原子数1~10的亚烷基,
[0020]
基d表示下述式(2)所示的碳原子数1~15的有机基,
[0021]
n表示1~5的数,
[0022][0023]
(式(2)中,r1、r2、r3各自独立地为氟原子、或者直链、支链、或环状的烷基,r1、r2、r3中的任意两者可以相互结合而形成环。)。]
[0024]
[2]根据[1]所述的纳米压印用抗蚀剂下层膜形成用组合物,基d为叔丁基、或三氟甲基。
[0025]
[3]根据[1]或[2]所述的纳米压印用抗蚀剂下层膜形成用组合物,基a中的具有芳香族环、稠合芳香族环、或稠合芳香族杂环的有机基为具有1个或多个苯环、萘环、蒽环、芘环、或者苯环与杂环或脂肪族环形成的稠环的有机基。
[0026]
[4]根据[1]~[3]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物,基a中的具有芳香族环、稠合芳香族环、或稠合芳香族杂环的有机基为可以在环上、环内、或环间包含选自n、s和o中的至少1种杂原子的碳原子数6~30的有机基。
[0027]
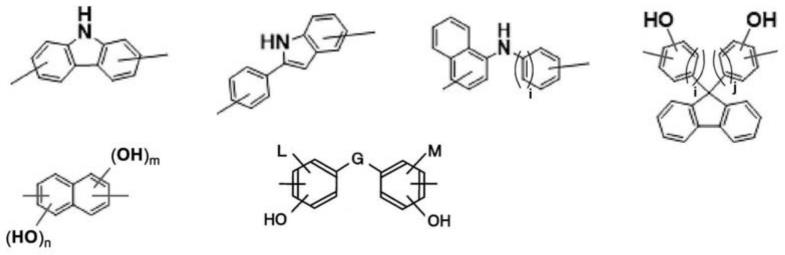
[5]根据[1]~[4]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物,基a为选自下述基团中的至少1种。
[0028][0029]
(式中,i、j、m、n各自独立地为1或2。g表示直接键合、或下述式中的任意者。
[0030]-ch2‑‑
ch(ch3)
‑‑
c(ch3)2‑‑
c(cf3)2‑‑
c(ch3)(c2h5)
‑‑
c(ch3)(c6h5)
‑‑
c(c6h5)2‑‑
so
2-[0031][0032]
l、m各自独立地表示氢原子、苯基、或c
1-3
烷基。)
[0033]
[6]根据[1]~[5]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物,基b为亚苯基、亚联苯基、亚萘基、亚蒽基、亚菲基。
[0034]
[7]根据[1]~[6]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物,基e为单键、或碳原子数1~6的直链亚烷基。
[0035]
[8]根据[1]~[7]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物,基e为单键。
[0036]
[9]根据[1]~[8]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物,在240℃进行了烧成时显示76
°
以上的对纯水的接触角,并且,在350℃进行了烧成时显示70
°
以上的对纯水的接触角。
[0037]
[10]根据[1]~[9]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物,其进一步包含交联剂。
[0038]
[11]根据[1]~[10]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物,其进一步包含选自酸、其盐和产酸剂中的至少一种。
[0039]
[12]根据[10]或[11]所述的纳米压印用抗蚀剂下层膜形成用组合物,在350℃进行了烧成时显示65
°
以上的对纯水的接触角。
[0040]
[13]一种抗蚀剂下层膜,其是由[1]~[12]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物形成的涂布膜的固化物。
[0041]
[14]一种抗蚀剂下层膜的制造方法,其包含:将[1]~[12]中任一项所述的纳米压
印用抗蚀剂下层膜形成用组合物涂布在半导体基板上并进行烧成。
[0042]
[15]一种图案形成方法,其包含下述工序:
[0043]
在半导体基板上由[1]~[12]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物形成抗蚀剂下层膜的工序;
[0044]
在上述抗蚀剂下层膜上应用固化性组合物的工序;
[0045]
使上述固化性组合物与模具接触的工序;
[0046]
向上述固化性组合物照射光或电子射线而制成固化膜的工序;以及
[0047]
将上述固化膜与上述模具分离的工序。
[0048]
[16]根据[15]所述的图案形成方法,在上述抗蚀剂下层膜上应用固化性组合物的工序包含:
[0049]
通过涂布或蒸镀而在上述抗蚀剂下层膜上形成密合层和/或包含99质量%以下、或50质量%以下的si的有机硅层,在该密合层和/或包含99质量%以下、或50质量%以下的si的有机硅层上应用固化性组合物。
[0050]
[17]一种半导体装置的制造方法,其包含下述工序:
[0051]
在半导体基板上由[1]~[12]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物形成抗蚀剂下层膜的工序;
[0052]
在该抗蚀剂下层膜上形成抗蚀剂膜的工序;
[0053]
通过光或电子射线的照射与显影而形成抗蚀剂图案的工序;
[0054]
通过所形成的抗蚀剂图案对该下层膜进行蚀刻的工序;以及
[0055]
通过被图案化了的下层膜对半导体基板进行加工的工序。
[0056]
[18]一种半导体装置的制造方法,其包含下述工序:
[0057]
在半导体基板上由[1]~[12]中任一项所述的纳米压印用抗蚀剂下层膜形成用组合物形成抗蚀剂下层膜的工序;
[0058]
在该抗蚀剂下层膜上形成硬掩模的工序;
[0059]
进一步在该硬掩模上形成抗蚀剂膜的工序;
[0060]
通过光或电子射线的照射与显影而形成抗蚀剂图案的工序;
[0061]
通过所形成的抗蚀剂图案对硬掩模进行蚀刻的工序;
[0062]
通过被图案化了的硬掩模对该下层膜进行蚀刻的工序;以及
[0063]
通过被图案化了的抗蚀剂下层膜对半导体基板进行加工的工序。
[0064]
发明的效果
[0065]
本发明涉及的酚醛清漆树脂不限于低温烧成时,在高温烧成时也显示异常高的纯水接触角(=疏水性)。此外,本发明涉及的酚醛清漆树脂在将交联剂、酸催化剂和表面活性剂混合、制成了材料时也在高温烧成时显示异常高的纯水接触角(=疏水性)。由此,能够提高与疏水性的上层膜的密合性,此外可以期待对疏水性气体显示良好的透过性。进一步,本发明涉及的酚醛清漆树脂显示良好的平坦化性,可以通过变更分子骨架来调整为适应于工艺的光学常数、蚀刻速度。
具体实施方式
[0066]
[纳米压印用抗蚀剂下层膜形成用组合物]
[0067]
本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物包含具有下述式(1)所示的重复单元结构的酚醛清漆树脂,任意选择地包含溶剂、其它成分。
[0068]
以下依次说明。
[0069][0070]
[在式(1)中,
[0071]
基a表示具有芳香族环、稠合芳香族环、或稠合芳香族杂环的有机基,
[0072]
基b表示具有芳香族环、或稠合芳香族环的有机基,
[0073]
基e表示单键、或者可以被取代且可以包含醚键和/或羰基的支链或直链的碳原子数1~10的亚烷基,
[0074]
基d表示下述式(2)所示的碳原子数1~15的有机基,
[0075]
n表示1~5的数。
[0076][0077]
(式中,r1、r2、r3各自独立地为氟原子、或者直链、支链、或环状的烷基,r1、r2、r3中的任意两者可以相互结合而形成环。)]
[0078]
[具有式(1)所示的重复单元结构的酚醛清漆树脂]
[0079]
基a和基b中的所谓“具有芳香族环的有机基”,是指具有以单环显示芳香族性的烃的基团。可举出例如,来源于苯、环辛四烯、以及具有任意的取代基的甲苯、二甲苯、均三甲苯、异丙基苯、苯乙烯的基团。进一步,也包含具有苯那样的芳香族环与环己烷、环己烯、甲基环己烷、甲基环己烯那样的脂肪族环形成的稠环的有机基、具有苯那样的芳香族环与呋喃、噻吩、吡咯、咪唑、吡喃、吡啶、嘧啶、吡嗪、吡咯烷、哌啶、哌嗪、吗啉那样的杂环形成的稠环的有机基。
[0080]
基a和基b中的所谓“具有稠合芳香族环的有机基”,是指具有以稠环显示芳香族性的烃的基团。可举出例如,来源于茚、萘、薁、蒽、菲、并四苯、苯并[9,10]菲、芘、的基团。
[0081]
基a中的所谓“具有稠合芳香族杂环的有机基”,是指具有以稠环显示芳香族性,并包含杂原子的烃的基团。可举出例如,来源于吲哚、嘌呤、喹啉、异喹啉、色烯、噻蒽、吩噻嗪、吩嗪、呫吨、吖啶、吩嗪、咔唑的基团。
[0082]
上述芳香族环、稠合芳香族环、和稠合芳香族杂环可以相互通过亚烷基等连接。
[0083]
优选地基a中的具有芳香族环、稠合芳香族环、或稠合芳香族杂环的有机基为碳原子数6~30的有机基。
[0084]
优选地基a中的具有芳香族环、稠合芳香族环、或稠合芳香族杂环的有机基为具有1个或多个苯环、萘环、或者苯环与杂环或脂肪族环形成的稠环的有机基。
[0085]
优选地基a中的具有芳香族环、稠合芳香族环、或稠合芳香族杂环的有机基为可以
在环上、环内、或环间包含选自n、s和o中的至少1种杂原子的碳原子数6~30的有机基。作为在环上包含的杂原子,可举出例如,氨基(例如,炔丙基氨基)、氰基所包含的氮原子、甲酰基、羟基、羧基、烷氧基(例如,炔丙基氧基)所包含的氧原子、硝基所包含的氮原子和氧原子。作为环内所包含的杂原子,可举出例如,呫吨所包含的氧原子、咔唑所包含的氮原子。作为环间所包含的杂原子,可举出-nh-键、-nhco-键、-o-键、-coo-键、-co-键、-s-键、-ss-键、-so
2-键所包含的氮原子、氧原子、硫原子。
[0086]
优选地基a为选自下述基团中的至少1种。
[0087][0088]
(式中,i、j、m、n各自独立地为1或2。g表示直接键合、或下述式的任意者。
[0089]-ch2‑‑
ch(ch3)
‑‑
c(ch3)2‑‑
c(cf3)2‑‑
c(ch3)(c2h5)
‑‑
c(ch3)(c6h5)
‑‑
c(c6h5)2‑‑
so
2-[0090][0091]
l、m各自独立地表示氢原子、苯基、或c
1-3
烷基。)
[0092]
优选地、基a为选自下述基团中的至少1种。
[0093][0094]
优选地基b为亚苯基、亚联苯基、亚萘基、亚蒽基、亚菲基。
[0095]
基d表示上述式(2)所示的碳原子数1~15的有机基,优选为碳原子数1~12、碳原子数1~10、碳原子数1~8、碳原子数1~6、碳原子数1~5、或碳原子数1~4的有机基。
[0096]
作为r1、r2、r3中的“直链、支链、或环状的烷基”,可举出例如,甲基、乙基、正丙基、异丙基、环丙基、正丁基、异丁基、仲丁基、叔丁基、环丁基、1-甲基-环丙基、2-甲基-环丙基、正戊基、1-甲基-正丁基、2-甲基-正丁基、3-甲基-正丁基、1,1-二甲基-正丙基、1,2-二甲基-正丙基、2,2-二甲基-正丙基、1-乙基-正丙基、环戊基、1-甲基-环丁基、2-甲基-环丁基、3-甲基-环丁基、1,2-二甲基-环丙基、2,3-二甲基-环丙基、1-乙基-环丙基、2-乙基-环丙基、正己基、1-甲基-正戊基、2-甲基-正戊基、3-甲基-正戊基、4-甲基-正戊基、1,1-二甲基-正丁基、1,2-二甲基-正丁基、1,3-二甲基-正丁基、2,2-二甲基-正丁基、2,3-二甲基-正丁基、3,3-二甲基-正丁基、1-乙基-正丁基、2-乙基-正丁基、1,1,2-三甲基-正丙基、1,2,2-三甲基-正丙基、1-乙基-1-甲基-正丙基、1-乙基-2-甲基-正丙基、环己基、1-甲基-环戊基、2-甲基-环戊基、3-甲基-环戊基、1-乙基-环丁基、2-乙基-环丁基、3-乙基-环丁基、1,2-二甲基-环丁基、1,3-二甲基-环丁基、2,2-二甲基-环丁基、2,3-二甲基-环丁基、2,4-二甲基-环丁基、3,3-二甲基-环丁基、1-正丙基-环丙基、2-正丙基-环丙基、1-异丙基-环丙基、2-异丙基-环丙基、1,2,2-三甲基-环丙基、1,2,3-三甲基-环丙基、2,2,3-三甲基-环丙基、1-乙基-2-甲基-环丙基、2-乙基-1-甲基-环丙基、2-乙基-2-甲基-环丙基、和2-乙基-3-甲基-环丙基。进一步,r1、r2、r3中的任意两者可以相互结合而形成环。
[0097]
优选地基d为叔丁基、或三氟甲基。
[0098]
基e为单键、或碳原子数1~6的直链亚烷基。作为直链亚烷基,可举出例如,亚甲基、亚乙基、亚丙基、亚丁基、亚戊基、亚己基。优选地基e为单键。
[0099]
n为1-5、1-4、或1-3的数,优选为1、2、3、4、或5,更优选为1、2、3、或4,最优选为1、2、或3。
[0100]
[合成方法]
[0101]
具有式(1)所示的重复单元结构的酚醛清漆树脂可以通过公知的方法来调制。例如,可以通过使h-a-h所示的含环化合物与ohc-b-e-d所示的醛化合物缩合来调制(式中,a、b、e、d与上述含义相同)。含环化合物、醛化合物都可以使用1种,也可以组合使用2种以上。在该缩合反应中,相对于含环化合物1摩尔,可以以0.1~10摩尔、优选为0.1~2摩尔的比例使用醛化合物。
[0102]
作为在缩合反应中使用的催化剂,可以使用例如硫酸、磷酸、高氯酸等无机酸类、对甲苯磺酸、对甲苯磺酸一水合物、甲磺酸等有机磺酸类、甲酸、草酸等羧酸类。催化剂的使用量根据所使用的催化剂的种类不同而不同,但相对于含环化合物(在多种的情况下为它们的合计)100质量份,通常为0.001~10,000质量份,优选为0.01~1,000质量份,更优选为0.05~100质量份。
[0103]
缩合反应在无溶剂的条件下也进行,但通常使用溶剂进行。作为溶剂,只要是可以溶解反应底物,不阻碍反应的溶剂,就没有特别限定。可举出例如,1,2-二甲氧基乙烷、二甘醇二甲基醚、丙二醇单甲基醚、丙二醇单甲基醚乙酸酯、四氢呋喃、二烷等。缩合反应温度通常为40℃~200℃,优选为100℃~180℃。反应时间根据反应温度不同而不同,但通常为5分钟~50小时,优选为5分钟~24小时。
[0104]
具有式(1)所示的重复单元结构的酚醛清漆树脂的重均分子量通常为500-100,000,优选为600-80,000、800-60,000、或1,000-50,000。
[0105]
本发明涉及的具有式(1)所示的重复单元结构的酚醛清漆树脂不加入交联剂等添
加剂、溶解于溶剂而涂布在基板(硅晶片)上,在240℃进行了烧成时显示76
°
以上的对纯水的接触角,在350℃进行了显示烧成时70
°
以上的对纯水的接触角。
[0106]
[溶剂]
[0107]
本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物可以包含溶剂。该溶剂只要是可以溶解具有式(1)所示的重复单元结构的酚醛清漆树脂、和根据需要添加的任意成分的溶剂,就没有特别限定。特别是,本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物由于以均匀的溶液状态使用,因此如果考虑其涂布性能,则推荐并用在光刻工序中一般使用的溶剂。
[0108]
作为那样的溶剂,可以举出例如,甲基溶纤剂乙酸酯、乙基溶纤剂乙酸酯、丙二醇、丙二醇单甲基醚、丙二醇单乙基醚、丙二醇单丙基醚、甲基异丁基甲醇、丙二醇单丁基醚、丙二醇单甲基醚乙酸酯、丙二醇单乙基醚乙酸酯、丙二醇单丙基醚乙酸酯、丙二醇单丁基醚乙酸酯、甲苯、二甲苯、甲基乙基酮、环戊酮、环己酮、2-羟基丙酸乙酯、2-羟基-2-甲基丙酸乙酯、乙氧基乙酸乙酯、羟基乙酸乙酯、2-羟基-3-甲基丁酸甲酯、3-甲氧基丙酸甲酯、3-甲氧基丙酸乙酯、3-乙氧基丙酸乙酯、3-乙氧基丙酸甲酯、丙酮酸甲酯、丙酮酸乙酯、乙二醇单甲基醚、乙二醇单乙基醚、乙二醇单丙基醚、乙二醇单丁基醚、乙二醇单甲基醚乙酸酯、乙二醇单乙基醚乙酸酯、乙二醇单丙基醚乙酸酯、乙二醇单丁基醚乙酸酯、二甘醇二甲基醚、二甘醇二乙基醚、二甘醇二丙基醚、二甘醇二丁基醚、丙二醇单甲基醚、丙二醇二甲基醚、丙二醇二乙基醚、丙二醇二丙基醚、丙二醇二丁基醚、乳酸乙酯、乳酸丙酯、乳酸异丙酯、乳酸丁酯、乳酸异丁酯、甲酸甲酯、甲酸乙酯、甲酸丙酯、甲酸异丙酯、甲酸丁酯、甲酸异丁酯、甲酸戊酯、甲酸异戊酯、乙酸甲酯、乙酸乙酯、乙酸戊酯、乙酸异戊酯、乙酸己酯、丙酸甲酯、丙酸乙酯、丙酸丙酯、丙酸异丙酯、丙酸丁酯、丙酸异丁酯、丁酸甲酯、丁酸乙酯、丁酸丙酯、丁酸异丙酯、丁酸丁酯、丁酸异丁酯、羟基乙酸乙酯、2-羟基-2-甲基丙酸乙酯、3-甲氧基-2-甲基丙酸甲酯、2-羟基-3-甲基丁酸甲酯、甲氧基乙酸乙酯、乙氧基乙酸乙酯、3-甲氧基丙酸甲酯、3-乙氧基丙酸乙酯、3-甲氧基丙酸乙酯、3-甲氧基丁基乙酸酯、3-甲氧基丙基乙酸酯、3-甲基-3-甲氧基丁基乙酸酯、3-甲基-3-甲氧基丁基丙酸酯、3-甲基-3-甲氧基丁基丁酸酯、乙酰乙酸甲酯、甲苯、二甲苯、甲基乙基酮、甲基丙基酮、甲基丁基酮、2-庚酮、3-庚酮、4-庚酮、环己酮、n,n-二甲基甲酰胺、n-甲基乙酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、4-甲基-2-戊醇、和γ-丁内酯等。这些溶剂可以单独使用,或以二种以上的组合而使用。
[0109]
它们之中,更优选为丙二醇单甲基醚、丙二醇单乙基醚、丙二醇单丙基醚、丙二醇单甲基醚乙酸酯、丙二醇单乙基醚乙酸酯、丙二醇单丙基醚乙酸酯,进一步优选为丙二醇单甲基醚、丙二醇单甲基醚乙酸酯。
[0110]
[交联剂]
[0111]
本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物可以包含交联剂。作为该交联剂,可举出三聚氰胺系、取代脲系、或它们的聚合物系等。优选为具有至少2个交联形成取代基的交联剂,为甲氧基甲基化甘脲(例如,四甲氧基甲基甘脲)、丁氧基甲基化甘脲、甲氧基甲基化三聚氰胺、丁氧基甲基化三聚氰胺、甲氧基甲基化苯胍胺、丁氧基甲基化苯胍胺、甲氧基甲基化脲、丁氧基甲基化脲、或甲氧基甲基化硫脲等化合物。此外,也可以使用这些化合物的缩合物。
[0112]
此外,作为上述交联剂,可以使用耐热性高的交联剂。作为耐热性高的交联剂,可
以优选使用在分子内含有具有芳香族环(例如,苯环、萘环)的交联形成取代基的化合物。
[0113]
该化合物可举出具有下述式(4)的部分结构的化合物、具有下述式(5)的重复单元的聚合物或低聚物。
[0114][0115]
上述r
11
、r
12
、r
13
、和r
14
为氢原子或碳原子数1~10的烷基,这些烷基可以使用上述例示。n1为1~4的整数,n2为1~(5-n1)的整数,(n1 n2)表示2~5的整数。n3为1~4的整数,n4为0~(4-n3),(n3 n4)表示1~4的整数。低聚物和聚合物可以在重复单元结构的数目为2~100、或2~50的范围内使用。
[0116]
以下例示式(4)和式(5)的化合物、聚合物、低聚物。
[0117]
[0118][0119]
上述化合物可以作为旭有机材工业株式会社、本州化学工业株式会社的制品而获得。例如,在上述交联剂中,式(4-23)的化合物可以作为本州化学工业株式会社、商品名tmom-bp而获得,式(4-24)的化合物可以作为旭有机材工业株式会社、商品名tm-bip-a而获得,式(4-28)的化合物可以作为商品名pgme-bip-a而获得。
[0120]
交联剂的添加量根据所使用的涂布溶剂、所使用的基板、所要求的溶液粘度、所要求的膜形状等变动而变动,但相对于全部固体成分为0.001质量%以上、0.01质量%以上、0.05质量%以上、0.5质量%以上、或1.0质量%以上,且为80质量%以下、50质量%以下、40质量%以下、20质量%以下、或10质量%以下。这些交联剂有时也发生由自缩合引起的交联反应,但在本发明的上述聚合物中存在交联性取代基的情况下,可以与这些交联性取代基发生交联反应。
[0121]
[酸和/或其盐和/或产酸剂]
[0122]
本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物可以包含酸和/或其盐和/或产酸剂。
[0123]
作为酸,可举出例如,对甲苯磺酸、三氟甲磺酸、水杨酸、5-磺基水杨酸、4-苯酚磺酸、樟脑磺酸、4-氯苯磺酸、苯二磺酸、1-萘磺酸、柠檬酸、苯甲酸、羟基苯甲酸、萘甲酸等。
[0124]
作为盐,也可以使用上述酸的盐。作为盐,没有限定,可以适合使用三甲基胺盐、三乙胺盐等氨衍生物盐、吡啶衍生物盐、吗啉衍生物盐等。
[0125]
酸和/或其盐可以仅使用一种,或可以组合使用二种以上。混配量相对于全部固体成分,通常为0.0001~20质量%,优选为0.0005~10质量%,进一步优选为0.01~5质量%。
[0126]
作为产酸剂,可举出热产酸剂、光产酸剂。
[0127]
作为热产酸剂,可举出2,4,4,6-四溴环己二烯酮、苯偶姻甲苯磺酸酯、2-硝基苄基甲苯磺酸酯、k-pure〔注册商标〕cxc-1612、k-pure cxc-1614、k-pure tag-2172、k-pure tag-2179、k-pure tag-2678、k-pure tag2689、k-pure tag2700(king industries社制)、和si-45、si-60、si-80、si-100、si-110、si-150(三新化学工业(株)制)以及有机磺酸烷基酯等。
[0128]
光产酸剂在抗蚀剂的曝光时产生酸。因此,可以调整下层膜的酸度。这是用于使下层膜的酸度与上层的抗蚀剂的酸度一致的一种方法。此外,通过下层膜的酸度的调整,可以调整形成在上层的抗蚀剂的图案形状。
[0129]
作为本发明的纳米压印用抗蚀剂下层膜形成用组合物所包含的光产酸剂,可举出盐化合物、磺酰亚胺化合物、和二磺酰基重氮甲烷化合物等。
[0130]
作为盐化合物,可举出二苯基碘六氟磷酸盐、二苯基碘三氟甲烷磺酸盐、二苯基碘九氟正丁烷磺酸盐、二苯基碘全氟正辛烷磺酸盐、二苯基碘樟脑磺酸盐、双(4-叔丁基苯基)碘樟脑磺酸盐和双(4-叔丁基苯基)碘三氟甲烷磺酸盐等碘盐化合物、以及三苯基锍六氟锑酸盐、三苯基锍九氟正丁烷磺酸盐、三苯基锍樟脑磺酸盐和三苯基锍三氟甲烷磺酸盐等锍盐化合物等。
[0131]
作为磺酰亚胺化合物,可举出例如n-(三氟甲磺酰氧基)琥珀酰亚胺、n-(九氟正丁烷磺酰氧基)琥珀酰亚胺、n-(樟脑磺酰氧基)琥珀酰亚胺和n-(三氟甲磺酰氧基)萘二甲酰亚胺等。
[0132]
作为二磺酰基重氮甲烷化合物,可举出例如,双(三氟甲基磺酰基)重氮甲烷、双(环己基磺酰基)重氮甲烷、双(苯基磺酰基)重氮甲烷、双(对甲苯磺酰基)重氮甲烷、双(2,4-二甲基苯磺酰基)重氮甲烷、和甲基磺酰基-对甲苯磺酰基重氮甲烷等。
[0133]
产酸剂可以仅使用一种,或可以组合使用二种以上。
[0134]
在使用产酸剂的情况下,作为其比例,相对于纳米压印用抗蚀剂下层膜形成用组合物的固体成分100质量份,为0.01~10质量份、或0.1~8质量份、或0.5~5质量份。
[0135]
本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物可以含有上述以外的任意成分。以下对各成分进行说明。
[0136]
[其它成分]
[0137]
在本发明的纳米压印用抗蚀剂下层膜形成用组合物中,为了不产生针孔、条痕等,进一步提高对不平表面的涂布性,可以混配表面活性剂。作为表面活性剂,可以举出例如聚氧乙烯月桂基醚、聚氧乙烯硬脂基醚、聚氧乙烯鲸蜡基醚、聚氧乙烯油基醚等聚氧乙烯烷基醚类、聚氧乙烯辛基苯酚醚、聚氧乙烯壬基苯酚醚等聚氧乙烯烷基芳基醚类、聚氧乙烯/聚
氧丙烯嵌段共聚物类、失水山梨糖醇单月桂酸酯、失水山梨糖醇单棕榈酸酯、失水山梨糖醇单硬脂酸酯、失水山梨糖醇单油酸酯、失水山梨糖醇三油酸酯、失水山梨糖醇三硬脂酸酯等失水山梨糖醇脂肪酸酯类、聚氧乙烯失水山梨糖醇单月桂酸酯、聚氧乙烯失水山梨糖醇单棕榈酸酯、聚氧乙烯失水山梨糖醇单硬脂酸酯、聚氧乙烯失水山梨糖醇三油酸酯、聚氧乙烯失水山梨糖醇三硬脂酸酯等聚氧乙烯失水山梨糖醇脂肪酸酯类等非离子系表面活性剂、
エフトップ
ef301、ef303、ef352(株式会社
トーケムプロダクツ
制,商品名)、
メガファック
f171、f173、r-40、r-40n、r-40lm(dic株式会社制,商品名)、
フロラード
fc430、fc431(住友
スリーエム
株式会社制,商品名)、
アサヒガード
ag710、
サーフロン
s-382、sc101、sc102、sc103、sc104、sc105、sc106(旭硝子株式会社制,商品名)等氟系表面活性剂、有机硅氧烷聚合物kp341(信越化学工业株式会社制)等。这些表面活性剂的混配量相对于抗蚀剂下层膜形成用组合物的全部固体成分通常为2.0质量%以下,优选为1.0质量%以下。这些表面活性剂可以单独使用,此外也可以以二种以上的组合使用。在使用表面活性剂的情况下,作为其比例,相对于纳米压印用抗蚀剂下层膜形成用组合物的固体成分100质量份为0.0001~5质量份、或0.001~1质量份、或0.01~0.5质量份。
[0138]
在本发明的纳米压印用抗蚀剂下层膜形成用组合物中,可以添加吸光剂、流变调节剂、粘接助剂等。流变调节剂对于提高下层膜形成用组合物的流动性而言是有效的。粘接助剂对于使半导体基板或抗蚀剂与下层膜的密合性提高而言是有效的。
[0139]
作为吸光剂,可以适合使用例如,“工業用色素
の
技術
と
市場(工业用色素的技术与市场)”(cmc出版)、“染料便覧(染料便览)”(有机合成化学协会编)所记载的市售的吸光剂,例如,c.i.分散黄1、3、4、5、7、8、13、23、31、49、50、51、54、60、64、66、68、79、82、88、90、93、102、114和124;c.i.分散橙1、5、13、25、29、30、31、44、57、72和73;c.i.分散红1、5、7、13、17、19、43、50、54、58、65、72、73、88、117、137、143、199和210;c.i.分散紫43;c.i.分散蓝96;c.i.荧光增白剂112、135和163;c.i.溶剂橙2和45;c.i.溶剂红1、3、8、23、24、25、27和49;c.i.颜料绿10;c.i.颜料棕2等。上述吸光剂通常相对于纳米压印用抗蚀剂下层膜形成用组合物的全部固体成分以10质量%以下、优选以5质量%以下的比例混配。
[0140]
流变调节剂主要出于提高纳米压印用抗蚀剂下层膜形成用组合物的流动性,特别是在烘烤工序中,提高抗蚀剂下层膜的膜厚均匀性、提高纳米压印用抗蚀剂下层膜形成用组合物向孔穴内部的填充性的目的添加。作为具体例,可以举出苯二甲酸二甲酯、苯二甲酸二乙酯、苯二甲酸二异丁酯、苯二甲酸二己酯、苯二甲酸丁基异癸基酯等苯二甲酸衍生物、己二酸二正丁酯、己二酸二异丁酯、己二酸二异辛酯、己二酸辛基癸基酯等己二酸衍生物、马来酸二正丁酯、马来酸二乙酯、马来酸二壬酯等马来酸衍生物、油酸甲酯、油酸丁酯、油酸四氢糠酯等油酸衍生物、或硬脂酸正丁酯、硬脂酸甘油酯等硬脂酸衍生物。这些流变调节剂相对于纳米压印用抗蚀剂下层膜形成用组合物的全部固体成分,通常以小于30质量%的比例混配。
[0141]
粘接助剂主要出于用于使基板或抗蚀剂与纳米压印用抗蚀剂下层膜形成用组合物的密合性提高,特别是在显影中不使抗蚀剂剥离的目的添加。作为具体例,可以举出三甲基氯硅烷、二甲基羟甲基氯硅烷、甲基二苯基氯硅烷、氯甲基二甲基氯硅烷等氯硅烷类、三甲基甲氧基硅烷、二甲基二乙氧基硅烷、甲基二甲氧基硅烷、二甲基羟甲基乙氧基硅烷、二苯基二甲氧基硅烷、苯基三乙氧基硅烷等烷氧基硅烷类、六甲基二硅氮烷、n,n
’‑
双(三甲基
甲硅烷基)脲、二甲基三甲基甲硅烷基胺、三甲基甲硅烷基咪唑等硅氮烷类、羟甲基三氯硅烷、γ-氯丙基三甲氧基硅烷、γ-氨基丙基三乙氧基硅烷、γ-环氧丙氧基丙基三甲氧基硅烷等硅烷类、苯并三唑、苯并咪唑、吲唑、咪唑、2-巯基苯并咪唑、2-巯基苯并噻唑、2-巯基苯并唑、尿唑、硫尿嘧啶、巯基咪唑、巯基嘧啶等杂环式化合物、1,1-二甲基脲、1,3-二甲基脲等脲、或硫脲化合物。这些粘接助剂相对于纳米压印用抗蚀剂下层膜形成用组合物的全部固体成分通常以小于5质量%,优选以小于2质量%的比例混配。
[0142]
本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物的固体成分通常为0.1~70质量%,优选为0.1~60质量%。固体成分为从纳米压印用抗蚀剂下层膜形成用组合物中除去了溶剂后的全部成分的含有比例。固体成分中的上述聚合物的比例按照1~100质量%、1~99.9质量%、50~99.9质量%、50~95质量%、50~90质量%的顺序优选。
[0143]
评价纳米压印用抗蚀剂下层膜形成用组合物是否为均匀的溶液状态的尺度之一是观察特定的微型过滤器的通过性,本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物能够通过孔径0.1μm的微型过滤器,呈现均匀的溶液状态。
[0144]
作为上述微型过滤器材质,可举出ptfe(聚四氟乙烯)、pfa(四氟乙烯/全氟烷基乙烯基醚共聚物)等氟系树脂、pe(聚乙烯)、upe(超高分子量聚乙烯)、pp(聚丙烯)、psf(聚砜)、pes(聚醚砜)、尼龙,但优选为ptfe(聚四氟乙烯)制。
[0145]
本发明涉及的具有式(1)所示的重复单元结构的酚醛清漆树脂混配溶剂和其它任意成分而制成纳米压印用抗蚀剂下层膜形成用组合物,涂布在基板(硅晶片)上,在350℃进行了烧成时显示65
°
以上的对纯水的接触角。
[0146]
以下,对使用了本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物的抗蚀剂下层膜的制造方法、图案形成方法和半导体装置的制造方法进行说明。
[0147]
[纳米压印用抗蚀剂下层膜的制造方法]
[0148]
在半导体装置的制造所使用的基板(例如,硅晶片基板、硅/二氧化硅被覆基板、氮化硅基板、玻璃基板、ito基板、聚酰亚胺基板、和低介电常数材料(low-k材料)被覆基板等)上,使用旋涂器、涂布机等适当的涂布方法而涂布本发明的纳米压印用抗蚀剂下层膜形成用组合物,然后,进行烧成从而形成抗蚀剂下层膜。作为进行烧成的条件,从烧成温度80℃~400℃、烧成时间0.3~60分钟中适当选择。优选为烧成温度150℃~350℃,烧成时间0.5~2分钟。这里,作为所形成的下层膜的膜厚,例如,为10~1000nm,或为20~500nm,或为30~400nm,或为50~300nm。此外,如果使用石英基板作为基板,则可以制作石英压印模具的复制品(模具复制品)。
[0149]
此外,也可以在本发明涉及的纳米压印用抗蚀剂下层膜上通过涂布或蒸镀而形成密合层和/或包含99质量%以下、或50质量%以下的si的有机硅层。例如,除了通过旋转涂布而形成日本特开2013-202982号公报、日本专利第5827180号公报所记载的密合层、wo2009/104552a1所记载的含有硅的抗蚀剂下层膜(无机抗蚀剂下层膜)形成用组合物的方法以外,可以通过cvd法等形成si系的无机材料膜。
[0150]
此外,将本发明涉及的纳米压印用抗蚀剂下层膜形成用组合物涂布在具备具有高低差的部分和不具有高低差的部分的半导体基板(所谓高低差基板)上,进行烧成,从而可以形成该具有高低差的部分和不具有高低差的部分的高低差为3~70nm的范围内的、抗蚀剂下层膜。
[0151]
[图案形成方法]
[0152]
本发明涉及的图案形成方法包含下述工序:
[0153]
在通过本发明涉及的抗蚀剂下层膜的制造方法而形成的抗蚀剂下层膜上应用固化性组合物的工序;
[0154]
使上述固化性组合物与模具接触的工序;
[0155]
向上述固化性组合物照射光或电子射线而制成固化膜的工序;以及
[0156]
将上述固化膜与上述模具分离的工序。
[0157]
[固化性组合物]
[0158]
作为在抗蚀剂下层膜上形成的光致抗蚀剂,只要是对曝光所使用的光感光的物质,就没有特别限定。负型光致抗蚀剂和正型光致抗蚀剂都可以使用。有由酚醛清漆树脂和1,2-萘醌重氮基磺酸酯构成的正型光致抗蚀剂、由具有通过酸进行分解而使碱溶解速度上升的基团的粘合剂和光产酸剂构成的化学放大型光致抗蚀剂、由通过酸进行分解而使光致抗蚀剂的碱溶解速度上升的低分子化合物和碱溶性粘合剂和光产酸剂构成的化学放大型光致抗蚀剂、以及由具有通过酸进行分解而使碱溶解速度上升的基团的粘合剂和通过酸进行分解而使光致抗蚀剂的碱溶解速度上升的低分子化合物和光产酸剂构成的化学放大型光致抗蚀剂等。可举出例如,
シプレー
社制商品名apex-e、住友化学工业株式会社制商品名par710、和信越化学工业株式会社制商品名sepr430等。此外,可以举出例如,proc.spie,vol.3999,330-334(2000)、proc.spie,vol.3999,357-364(2000)、proc.spie,vol.3999,365-374(2000)所记载那样的、含氟原子聚合物系光致抗蚀剂。
[0159]
[应用固化性组合物的工序]
[0160]
本工序为在通过本发明涉及的抗蚀剂下层膜的制造方法而形成的抗蚀剂下层膜上应用固化性组合物的工序。作为应用固化性组合物的方法,可以使用例如,喷墨法、浸渍涂布法、气刀涂布法、帘式涂布法、线棒涂布法、凹版涂布法、挤压涂布法、旋转涂布法、狭缝扫描法等。为了将固化性组合物作为液滴而应用,喷墨法是适合的,为了涂布固化性组合物,旋转涂布法是适合的。在本工序中,也可以在抗蚀剂下层膜上通过涂布或蒸镀而形成密合层和/或包含99质量%以下、或50质量%以下的si的有机硅层,在其上应用固化性组合物。
[0161]
[使固化性组合物与模具接触的工序]
[0162]
在本工序中,使固化性组合物与模具接触。例如,如果使作为液体的固化性组合物、与具有用于转印图案形状的原型图案的模具接触,则形成固化性组合物被填充于模具表面的微细图案的凹部的液膜。
[0163]
考虑后述照射光或电子射线的工序,推荐使用以光透过性材料作为基材的模具。具体而言,模具基材优选为玻璃、石英、pmma、聚碳酸酯树脂等光透明性树脂、透明金属蒸镀膜、聚二甲基硅氧烷等柔软膜、光固化膜、金属膜等。从热膨胀系数小而图案偏斜小考虑,模具基材更优选为石英。
[0164]
模具在表面所具有的微细图案优选具有4nm以上、200nm以下的图案高度。为了提升基板的加工精度,需要某种程度的图案高度,图案高度低时,在后述将固化膜与模具分离的工序中将模具从固化膜剥下的力小,此外,抗蚀剂图案被拉碎而残存在掩模侧的缺陷数少。考虑这些而推荐选择采用适当的平衡的图案高度。
[0165]
此外,也有时由于将模具剥下时的冲击引起的抗蚀剂图案的弹性变形而使相邻抗蚀剂图案彼此接触,抗蚀剂图案粘连或破损。这有时可以通过使图案高度相对于图案宽度为2倍左右以下(长宽比2以下)来避免。
[0166]
为了使固化性组合物与模具的表面的剥离性提高,也可以预先对模具进行表面处理。作为表面处理的方法,可举出在模具的表面涂布脱模剂而形成脱模剂层的方法。作为脱模剂,可举出有机硅系脱模剂、氟系脱模剂、烃系脱模剂、聚乙烯系脱模剂、聚丙烯系脱模剂、石蜡系脱模剂、褐煤系脱模剂、巴西棕榈(
カルナバ
)系脱模剂等。优选为氟系和烃系的脱模剂。作为市售品,有例如,
ダイキン
工業(株)制的
オプツール
(注册商标)dsx等。脱模剂可以单独使用一种,也可以并用二种以上。
[0167]
在本工序中,在使模具与固化性组合物接触时,施加于固化性组合物的压力没有特别限定。推荐0mpa以上、100mpa以下的压力。压力优选为0mpa以上,且为50mpa以下、30mpa以下、或20mpa以下。
[0168]
在前工序(应用固化性组合物的工序)中固化性组合物的液滴的预展开进行的情况下,本工序中的固化性组合物的展开迅速地完成。其结果,可以缩短使模具与固化性组合物接触的时间。进行接触的时间没有特别限定,但优选为0.1秒以上,且为600秒以下、3秒以下、或1秒以下。如果接触时间过短,则展开和装填变得不充分,可能会发生被称为未填充缺陷的缺陷。
[0169]
本工序在大气气氛下、减压气氛下、非活性气体气氛下的任一条件下都可以进行,但优选在0.0001气压以上、10气压以下的压力下进行。为了防止由氧、水分引起的对固化反应的影响,推荐在减压气氛下、或非活性气体气氛下进行。作为为了形成非活性气体气氛而可以使用的非活性气体的具体例,可举出氮气、二氧化碳、氦气、氩气、cfc、hcfc、hfc、或它们的混合气体。
[0170]
本工序可以在包含冷凝性气体的气氛(以下,称为“冷凝性气体气氛”)下进行。在本说明书中所谓冷凝性气体,是指与固化性组合物一起被填充在形成在模具上的微细图案的凹部和模具与基板的间隙时,由于在填充时产生的毛细管压力而冷凝、液化的气体。另外,冷凝性气体在本工序中在固化性组合物与模具接触前在气氛中作为气体而存在。如果在冷凝性气体气氛下进行本工序,则被填充在微细图案的凹部的气体由于由固化性组合物产生的毛细管压力而液化,从而气泡消失,因此填充性优异。冷凝性气体可以溶解于固化性组合物。
[0171]
冷凝性气体的沸点只要为本工序的气氛温度以下就没有限定,但优选为-10℃以上、或 10℃以上、 23℃以下。
[0172]
本工序的气氛温度下的冷凝性气体的蒸气压只要是模具压力以下,就没有特别限定。优选为0.1mpa~0.4mpa的范围。
[0173]
作为冷凝性气体,具体而言,可举出三氯氟甲烷等氯氟烃(cfc)、碳氟化合物(fc)、氢氯氟烃(hcfc)、1,1,1,3,3-五氟丙烷(chf2ch2cf3、hfc-245fa、pfp)等氢氟烃(hfc)、五氟乙基甲基醚(cf3cf2och3、hfe-245mc)等氢氟醚(hfe)等。
[0174]
冷凝性气体可以单独使用一种,也可以混合使用二种以上。此外,这些冷凝性气体可以与空气、氮气、二氧化碳、氦气、氩气等非冷凝性气体混合使用。作为与冷凝性气体混合的非冷凝性气体,优选为空气、氦气。
[0175]
[向固化性组合物照射光或电子射线而制成固化膜的工序]
[0176]
在本工序中,向固化性组合物照射光或电子射线而制成固化膜。即,向被填充于模具的微细图案的固化性组合物经由模具而照射光或电子射线,使被填充于模具的微细图案的固化性组合物在该状态下直接固化,从而制成具有图案形状的固化膜。
[0177]
光或电子射线根据固化性组合物的灵敏度波长来选择。具体而言,可以适当选择使用150nm以上且400nm以下的波长的紫外光、x射线、电子射线等。作为光或电子射线的光源,可举出例如,高压水银灯、超高压水银灯、低压水银灯、deep-uv灯、碳弧灯、化学灯、金属卤化物灯、氙灯、krf受激准分子激光器、arf受激准分子激光器、f2受激准分子激光器等。光源数可以为1个,也可以为多个。照射可以对被填充于模具的微细图案的固化性组合物的整体进行,也可以仅对一部分区域进行。光照射可以对基板上的全部区域间断地进行多次,也可以对全部区域连续照射。此外,也可以对基板上的一部分区域进行第一次照射,对与该区域不同的区域进行第二次照射。
[0178]
这样操作而获得的固化膜优选具有1nm以上、或10nm以上、且10mm以下、或100μm以下的尺寸的图案。
[0179]
[将固化膜与模具分离的工序]
[0180]
在本工序中,将固化膜与模具分离。将具有图案形状的固化膜与模具分离,具有成为形成在模具上的微细图案的反转图案的图案形状的固化膜在自立了的状态下获得。
[0181]
作为将具有图案形状的固化膜与模具分离的方法,只要是使固化膜与模具沿相对远离的方向移动的手段,只要具有图案形状的固化膜的一部分不会物理地破损,就没有特别限定,各种条件等也没有特别限定。例如,可以将基板固定而使模具以远离基板的方式移动而剥离,也可以将模具固定而使基板以远离模具的方式移动而剥离。或者,可以将基板与模具向相反方向拉伸使其移动而剥离。
[0182]
另外,在冷凝性气体气氛下进行了使上述固化性组合物与模具接触的工序的情况下,在本工序中在将固化膜与模具分离时,随着固化膜与模具接触的界面的压力降低而冷凝性气体气化。由此,可以使作为为了将固化膜与模具分离而需要的力的脱模力减少。
[0183]
通过以上工序,可以调制在所希望的位置具有来源于模具的凹凸形状的所希望的凹凸图案形状的固化膜。
[0184]
[半导体装置的制造方法]
[0185]
将通过本发明的图案形成方法而形成的光致抗蚀剂(上层)的图案作为保护膜而进行无机下层膜(中间层)的除去,接着以由被图案化了的光致抗蚀剂和无机下层膜(中间层)构成的膜作为保护膜,进行有机下层膜(下层)的除去。最后,以被图案化了的无机下层膜(中间层)和有机下层膜(下层)作为保护膜,进行半导体基板的加工。
[0186]
首先,将除去了光致抗蚀剂的部分的无机下层膜(中间层)通过干蚀刻而除去,使半导体基板露出。在无机下层膜的干蚀刻中可以使用四氟甲烷(cf4)、全氟环丁烷(c4f8)、全氟丙烷(c3f8)、三氟甲烷、一氧化碳、氩气、氧气、氮气、六氟化硫、二氟甲烷、三氟化氮和三氟化氯、氯气、三氯硼烷和二氯硼烷等气体。在无机下层膜的干蚀刻中优选使用卤素系气体,更优选采用氟系气体。作为氟系气体,可举出例如,四氟甲烷(cf4)、全氟环丁烷(c4f8)、全氟丙烷(c3f8)、三氟甲烷、和二氟甲烷(ch2f2)等。
[0187]
然后,以由被图案化了的光致抗蚀剂和无机下层膜构成的膜作为保护膜而进行有
机下层膜的除去。有机下层膜(下层)优选通过采用氧系气体的干蚀刻而进行。因为大量包含硅原子的无机下层膜通过采用氧系气体的干蚀刻不易被除去。
[0188]
最后,进行半导体基板的加工。半导体基板的加工优选通过采用氟系气体的干蚀刻而进行。
[0189]
作为氟系气体,可举出例如,四氟甲烷(cf4)、全氟环丁烷(c4f8)、全氟丙烷(c3f8)、三氟甲烷、和二氟甲烷(ch2f2)等。
[0190]
此外,可以在抗蚀剂下层膜的上层,在光致抗蚀剂的形成前形成有机系的防反射膜。作为这里使用的防反射膜组合物,没有特别限制,可以从迄今为止在光刻工艺中惯用的物质中任意选择而使用,此外,可以通过惯用的方法,例如,采用旋涂器、涂布机的涂布和烧成来进行防反射膜的形成。
[0191]
在本发明中可以在基板上形成了有机下层膜后,在其上成膜无机下层膜,进一步在其上被覆光致抗蚀剂。由此光致抗蚀剂的图案宽度变窄,即使在为了防止图案倒塌而薄薄地被覆了光致抗蚀剂的情况下,也能够通过选择适当的蚀刻气体而进行基板的加工。例如,能够将相对于光致抗蚀剂成为充分快的蚀刻速度的氟系气体作为蚀刻气体而对抗蚀剂下层膜进行加工,此外能够将相对于无机下层膜成为充分快的蚀刻速度的氟系气体作为蚀刻气体而进行基板的加工,进一步可以将相对于有机下层膜成为充分快的蚀刻速度的氧系气体作为蚀刻气体而进行基板的加工。
[0192]
此外,由抗蚀剂下层膜形成用组合物形成的抗蚀剂下层膜根据在光刻工艺中使用的光的波长,有时具有对该光的吸收。进而,在那样的情况下,可以作为具有防止来自基板的反射光的效果的防反射膜起作用。进一步,由本发明的抗蚀剂下层膜形成用组合物形成的下层膜也能够作为硬掩模起作用。本发明的下层膜也能够作为下述层而使用:用于防止基板与光致抗蚀剂的相互作用的层、具有防止光致抗蚀剂所使用的材料或在对光致抗蚀剂的曝光时生成的物质对基板的不良作用的功能的层、具有防止在加热烧成时从基板生成的物质向上层光致抗蚀剂扩散的功能的层、和用于使由半导体基板电介质层引起的光致抗蚀剂层的中毒效果减少的阻挡层等。
[0193]
此外,由抗蚀剂下层膜形成用组合物形成的下层膜被应用于在双镶嵌工艺中使用的形成了通孔的基板,可以作为能够没有间隙地填充孔穴的埋入材而使用。此外,也可以作为用于将具有凹凸的半导体基板的表面平坦化的平坦化材而使用。
[0194]
实施例
[0195]
接下来举出实施例等具体地说明本发明的内容,但本发明不限定于它们。
[0196]
在下述合成例1中获得的树脂(聚合物)的重均分子量为由凝胶渗透色谱(以下,简称为gpc。)得到的测定结果。测定使用東
ソー
株式会社制gpc装置,测定条件等如下所述。
[0197]
gpc柱:shodex kf803l、shodex kf802、shodex kf801〔注册商标〕(昭和电工株式会社)
[0198]
柱温度:40℃
[0199]
溶剂:四氢呋喃(thf)
[0200]
流量:1.0ml/分钟
[0201]
标准试样:聚苯乙烯(東
ソー
株式会社制)
[0202]
干蚀刻速度的测定所使用的蚀刻器和蚀刻气体使用了以下物质。
[0203]
rie-10nr(
サムコ
制):cf4[0204]
[合成例1]
[0205]
在100ml二口烧瓶中加入4-叔丁基苯甲醛(东京化成工业株式会社制)9.71g、咔唑(东京化成工业株式会社制)10.00g、丙二醇单甲基醚乙酸酯(以下,记载为pgmea)48.68g、甲磺酸(东京化成工业株式会社制)1.15g。然后加热直到150℃,回流搅拌30分钟。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-1)。通过gpc以聚苯乙烯换算测定的重均分子量mw为4,900。
[0206][0207]
[合成例2]
[0208]
在100ml二口烧瓶中加入4-(三氟甲基)-苯甲醛(东京化成工业株式会社制)10.42g、咔唑(东京化成工业株式会社制)10.00g、pgmea 50.34g、甲磺酸(东京化成工业株式会社制)1.15g。然后加热直到150℃,回流搅拌3.5小时。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-2)。
[0209]
通过gpc以聚苯乙烯换算测定的重均分子量mw为4,200。
[0210][0211]
[合成例3]
[0212]
在100ml二口烧瓶中加入4-叔丁基苯甲醛(东京化成工业株式会社制)8.39g、2-苯基吲哚(东京化成工业株式会社制)10.00g、pgmea 45.24g、甲磺酸(东京化成工业株式会社制)0.99g。然后加热直到150℃,回流搅拌17小时。反应结束后,将该溶液滴加到甲醇/水混合液中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-3)。通过gpc以聚苯乙烯换算测定的重均分子量mw为1,200。
[0213][0214]
[合成例4]
[0215]
在100ml二口烧瓶中加入4-(三氟甲基)-苯甲醛(东京化成工业株式会社制)9.01g、2-苯基吲哚(东京化成工业株式会社制)10.00g、pgmea 46.68g、甲磺酸(东京化成工业株式会社制)0.99g。然后加热直到150℃,回流搅拌17小时。反应结束后,将该溶液滴加到甲醇/水混合液中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-4)。通过gpc以聚苯乙烯换算测定的重均分子量mw为2,200。
[0216][0217]
[合成例5]
[0218]
在100ml二口烧瓶中加入4-叔丁基苯甲醛(东京化成工业株式会社制)7.40g、n-苯基-1-萘胺(东京化成工业株式会社制)10.00g、pgmea18.50g、甲磺酸(东京化成工业株式会社制)1.10g。然后加热直到150℃,回流搅拌10分钟。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-5)。通过gpc以聚苯乙烯换算测定的重均分子量mw为6,000。
[0219][0220]
[合成例6]
[0221]
在100ml二口烧瓶中加入4-(三氟甲基)-苯甲醛(东京化成工业株式会社制)7.95g、n-苯基-1-萘胺(东京化成工业株式会社制)10.00g、pgmea18.50g、甲磺酸(东京化成工业株式会社制)0.55g。然后加热直到150℃,回流搅拌10分钟。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-6)。通过gpc以聚苯乙烯换算测定的重均分子量mw为30,000。
[0222][0223]
[合成例7]
[0224]
在100ml二口烧瓶中加入4-叔丁基苯甲醛(东京化成工业株式会社制)4.63g、9,9-双(4-羟基苯基)芴(东京化成工业株式会社制)10.00g、pgmea22.77g、甲磺酸(东京化成工业株式会社制)0.55g。然后加热直到150℃,回流搅拌5.5小时。反应结束后,将该溶液滴加到甲醇/水混合液中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-7)。通过gpc以聚苯乙烯换算测定的重均分子量mw为2,000。
[0225][0226]
[合成例8]
[0227]
在100ml二口烧瓶中加入4-(三氟甲基)-苯甲醛(东京化成工业株式会社制)4.97g、9,9-双(4-羟基苯基)芴(东京化成工业株式会社制)10.00g、pgmea 23.28g、甲磺酸(东京化成工业株式会社制)0.55g。然后加热直到150℃,回流搅拌5.5小时。反应结束后,将该溶液滴加到甲醇/水混合液中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-8)。通过gpc以聚苯乙烯换算测定的重均分子量mw
为10,000。
[0228][0229]
[合成例9]
[0230]
在100ml二口烧瓶中加入4-叔丁基苯甲醛(东京化成工业株式会社制)10.13g、1,5-二羟基萘(东京化成工业株式会社制)10.00g、pgmea 49.77g、甲磺酸(东京化成工业株式会社制)1.20g。然后加热直到150℃,回流搅拌1小时45分钟。反应结束后,将该溶液滴加到甲醇/水混合液中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-9)。通过gpc以聚苯乙烯换算测定的重均分子量mw为5,200。
[0231][0232]
[合成例10]
[0233]
在100ml二口烧瓶中加入4-(三氟甲基)-苯甲醛(东京化成工业株式会社制)10.87g、1,5-二羟基萘(东京化成工业株式会社制)10.00g、pgmea51.50g、甲磺酸(东京化成工业株式会社制)1.20g。然后加热直到150℃,回流搅拌2小时15分钟。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-10)。通过gpc以聚苯乙烯换算测定的重均分子量mw为8,300。
[0234][0235]
[合成例11]
[0236]
在100ml二口烧瓶中加入4-叔丁基苯甲醛(东京化成工业株式会社制)4.69g、双酚m(东京化成工业株式会社制)10.00g、pgmea 35.56g、甲磺酸(东京化成工业株式会社制)0.56g。然后加热直到150℃,回流搅拌16小时。反应结束后,将该溶液滴加到甲醇/水混合液中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-11)。通过gpc以聚苯乙烯换算测定的重均分子量mw为8,000。
[0237][0238]
[合成例12]
[0239]
在100ml二口烧瓶中加入4-(三氟甲基)-苯甲醛(东京化成工业株式会社制)5.03g、双酚m(东京化成工业株式会社制)10.00g、pgmea 36.36g、甲磺酸(东京化成工业株式会社制)0.56g。然后加热直到150℃,回流搅拌5小时。反应结束后,将该溶液滴加到甲醇/
水混合液中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(2-12)。通过gpc以聚苯乙烯换算测定的重均分子量mw为6,500。
[0240][0241]
[比较合成例1]
[0242]
在100ml二口烧瓶中加入苯甲醛(东京化成工业株式会社制)6.35g、咔唑(东京化成工业株式会社制)10.00g、pgmea 40.84g、甲磺酸(东京化成工业株式会社制)1.15g。然后加热直到150℃,回流搅拌约30分钟。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(1-1)。通过gpc以聚苯乙烯换算测定的重均分子量mw为52,300。
[0243][0244]
[比较合成例2]
[0245]
在100ml二口烧瓶中加入苯甲醛(东京化成工业株式会社制)5.49g、2-苯基吲哚(东京化成工业株式会社制)10.00g、pgmea 16.49g、甲磺酸(东京化成工业株式会社制)0.99g。然后加热直到150℃,回流搅拌约5小时。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(1-2)。通过gpc以聚苯乙烯换算测定的重均分子量mw为1,600。
[0246][0247]
[比较合成例3]
[0248]
在100ml二口烧瓶中加入苯甲醛(东京化成工业株式会社制)4.84g、n-苯基-1-萘胺(东京化成工业株式会社制)10.00g、pgmea 36.67g、甲磺酸(东京化成工业株式会社制)0.88g。然后加热直到150℃,回流搅拌约15分钟。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(1-3)。通过gpc以聚苯乙烯换算测定的重均分子量mw为5,900。
[0249][0250]
[比较合成例4]
[0251]
在100ml二口烧瓶中加入苯甲醛(东京化成工业株式会社制)3.03g、9,9-双(4-羟基苯基)芴(东京化成工业株式会社制)10.00g、pgmea32.68g、甲磺酸(东京化成工业株式会社制)0.55g。然后加热直到150℃,回流搅拌约17.5小时。反应结束后,将该溶液滴加到甲醇
中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(1-4)。通过gpc以聚苯乙烯换算测定的重均分子量mw为10,300。
[0252][0253]
[比较合成例5]
[0254]
在100ml二口烧瓶中加入苯甲醛(东京化成工业株式会社制)6.62g、1,5-二羟基萘(东京化成工业株式会社制)10.00g、pgmea 41.58g、甲磺酸(东京化成工业株式会社制)1.20g。然后加热直到150℃,回流搅拌约1.5小时。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(1-5)。通过gpc以聚苯乙烯换算测定的重均分子量mw为5,300。
[0255][0256]
[比较合成例6]
[0257]
在100ml二口烧瓶中加入对甲苯甲醛(东京化成工业株式会社制)7.19g、咔唑(东京化成工业株式会社制)10.00g、pgmea 42.80g、甲磺酸(东京化成工业株式会社制)1.15g。然后加热直到150℃,回流搅拌约30分钟。反应结束后,将该溶液滴加到甲醇中,使其再沉淀。将所得的沉淀物进行抽滤,将滤物在60℃减压干燥一晚。所得的聚合物相当于式(1-6)。通过gpc以聚苯乙烯换算测定的重均分子量mw为5,900。
[0258][0259]
实施例所使用的原料的化学结构(例示)和简称如下所述。
[0260][0261]
[实施例1]
[0262]
将在合成例1中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μ
m的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0263]
[实施例2]
[0264]
将在合成例2中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0265]
[实施例3]
[0266]
将在合成例3中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0267]
[实施例4]
[0268]
将在合成例4中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0269]
[实施例5]
[0270]
将在合成例5中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0271]
[实施例6]
[0272]
将在合成例6中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0273]
[实施例7]
[0274]
将在合成例7中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0275]
[实施例8]
[0276]
将在合成例8中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0277]
[实施例9]
[0278]
将在合成例9中获得的树脂溶解于丙二醇单甲基醚(以下,记载为pgme)后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgme使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0279]
[实施例10]
[0280]
将在合成例10中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0281]
[实施例11]
[0282]
将在合成例11中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0283]
[实施例12]
[0284]
将在合成例12中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0285]
[实施例13]
[0286]
将在合成例1中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。在该树脂溶液2.65g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea 0.12g、tmom-bp(本州化学工业株式会社制)0.09g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme 0.45g、pgmea 4.34g、pgme 2.35g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0287]
[实施例14]
[0288]
将在合成例2中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为20.1质量%)。在该树脂溶液3.00g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea 0.12g、tmom-bp(本州化学工业株式会社制)0.09g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme 0.34g、pgmea 4.00g、pgme 2.49g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0289]
[实施例15]
[0290]
将在合成例3中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为19.0质量%)。在该树脂溶液1.94g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea 0.07g、pgme-bip-a(
ファインケム
株式会社制)0.18g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme 0.46g、pgmea 2.91g、pgme 1.43g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0291]
[实施例16]
[0292]
将在合成例4中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为20.8质量%)。在该树脂溶液2.02g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea 0.08g、tmom-bp(本州化学工业株式会社制)0.06g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme 0.31g、pgmea 2.88g、pgme 1.64g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0293]
[实施例17]
[0294]
将在合成例5中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为19.1质量%)。在该树脂溶液3.01g中加入含有1质量%表面活性剂(dic株式会社
制,
メガファック
r-40)的pgmea 0.12g、pgme-bip-a(
ファインケム
株式会社制)0.19g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme 0.43g、pgmea 3.96g、pgme 2.29g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0295]
[实施例18]
[0296]
将在合成例6中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为20.5质量%)。在该树脂溶液2.92g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea0.12g、tmom-bp(本州化学工业株式会社制)0.09g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme0.45g、pgmea4.07g、pgme2.35g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0297]
[实施例19]
[0298]
将在合成例7中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.2质量%)。在该树脂溶液2.56g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea0.11g、tmom-bp(本州化学工业株式会社制)0.11g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme0.85g、pgmea4.41g、pgme1.95g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0299]
[实施例20]
[0300]
将在合成例8中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.8质量%)。在该树脂溶液2.49g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea0.11g、tmom-bp(本州化学工业株式会社制)0.11g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme0.85g、pgmea4.47g、pgme1.95g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0301]
[实施例21]
[0302]
将在合成例9中获得的树脂溶解于pgme后,经过离子交换而获得了树脂溶液(固体成分为19.7质量%)。在该树脂溶液2.88g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea0.11g、tmom-bp(本州化学工业株式会社制)0.11g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme0.85g、pgmea2.68g、pgme3.36g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0303]
[实施例22]
[0304]
将在合成例10中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.9质量%)。在该树脂溶液2.48g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea0.11g、tmom-bp(本州化学工业株式会社制)0.11g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme0.85g、pgmea4.48g、
pgme1.95g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0305]
[实施例23]
[0306]
将在合成例11中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为17.9质量%)。在该树脂溶液1.93g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea0.07g、tmom-bp(本州化学工业株式会社制)0.07g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme0.26g、pgmea2.25g、pgme1.42g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0307]
[实施例24]
[0308]
将在合成例12中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为17.8质量%)。在该树脂溶液3.24g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea0.12g、tmom-bp(本州化学工业株式会社制)0.12g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme0.43g、pgmea3.73g、pgme2.37g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0309]
[比较例1]
[0310]
将在比较合成例1中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为18.4质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0311]
[比较例2]
[0312]
将在比较合成例2中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.5质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0313]
[比较例3]
[0314]
将在比较合成例3中获得的树脂溶解于环己酮后,经过离子交换而获得了树脂溶液(固体成分为17.7质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0315]
[比较例4]
[0316]
将在比较合成例4中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为15.9质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0317]
[比较例5]
[0318]
将在比较合成例5中获得的树脂溶解于pgme后,经过离子交换而获得了树脂溶液(固体成分为18.2质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0319]
[比较例6]
[0320]
将在比较合成例6中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液
(固体成分为26.0质量%)。以树脂固体成分成为5%的方式加入pgmea使其混合,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0321]
[比较例7]
[0322]
将在比较合成例6中获得的树脂溶解于pgmea后,经过离子交换而获得了树脂溶液(固体成分为22.6质量%)。在该树脂溶液2.19g中加入含有1质量%表面活性剂(dic株式会社制,
メガファック
r-40)的pgmea0.11g、tmom-bp(本州化学工业株式会社制)0.11g、含有2质量%吡啶对羟基苯磺酸盐(东京化成工业株式会社)的pgme0.85g、pgmea4.78g、pgme1.95g使其溶解,利用孔径0.1μm的聚四氟乙烯制微型过滤器进行过滤,调制出抗蚀剂下层膜形成用组合物的溶液。
[0323]
(聚合物的接触角测定)
[0324]
将实施例1-12和比较例1-6中调制的抗蚀剂下层膜形成用组合物的溶液分别使用旋转涂布机涂布在硅晶片上,在电热板上在240℃烧成60秒或在350℃烧成60秒,形成了聚合物膜。然后,使用协和界面科学株式会社制的接触角计,测定了聚合物对纯水的接触角。
[0325]
[表1]
[0326]
表1对纯水的接触角(
°
)
[0327]
[0328]
如上所述,具有叔丁基或三氟甲基的酚醛清漆树脂与类似的骨架相比,不仅低温烧成时,在高温烧成时也显示异常高的纯水接触角(=疏水性),明确具有优势性。在下个项目中,显示在具有叔丁基或三氟甲基的酚醛清漆树脂中混合交联剂、酸催化剂和表面活性剂、制成材料时的物性的评价结果。
[0329]
(材料的接触角测定)
[0330]
将在实施例13-24和比较例7中调制的抗蚀剂下层膜形成用组合物的溶液分别使用旋转涂布机涂布在硅晶片上,在电热板上在350℃烧成60秒,形成了200nm的抗蚀剂下层膜。然后,使用协和界面科学株式会社制的接触角计,测定了对纯水的接触角。
[0331]
[表2]
[0332]
表2对纯水的接触角(
°
)
[0333][0334]
如上所述,具有叔丁基或三氟甲基的酚醛清漆树脂在制成材料时,也在高温烧成时显示异常高的纯水接触角(=疏水性)。由此,能够提高与疏水性的上层膜的密合性,此外预想对疏水性气体显示良好的透过性。
[0335]
(在抗蚀剂溶剂中的溶出试验)
[0336]
将在实施例13-24和比较例7中调制的抗蚀剂下层膜形成用组合物的溶液分别使用旋转涂布机涂布在硅晶片上,在电热板上在350℃烧成60秒,形成了抗蚀剂下层膜(膜厚0.20μm)。将这些抗蚀剂下层膜浸渍于作为抗蚀剂所使用的溶剂的乳酸乙酯、丙二醇单甲基醚、丙二醇单甲基醚乙酸酯、和环己酮中。这些抗蚀剂下层膜不溶于这些溶剂。
[0337]
(光学常数测定)
[0338]
将在实施例13-24和比较例7中调制的抗蚀剂下层膜形成用组合物的溶液分别使
用旋转涂布机涂布在硅晶片上。在电热板上在350℃烧成60秒,形成了抗蚀剂下层膜(膜厚0.05μm)。将这些抗蚀剂下层膜使用光谱椭偏仪测定了波长193nm下的折射率(n值)和光学吸光系数(也称为k值、衰减系数)(表3)。
[0339]
[表3]
[0340]
表3折射率n和光学吸光系数k
[0341][0342]
如上所述,具有叔丁基或三氟甲基的酚醛清漆树脂可以通过变更分子骨架来调整为适应于工艺的光学常数。
[0343]
[干蚀刻速度的测定]
[0344]
将在实施例13-24和比较例7中调制的抗蚀剂下层膜形成用组合物的溶液分别使用旋转涂布机涂布在硅晶片上。在电热板上在350℃烧成60秒,形成了抗蚀剂下层膜(膜厚0.20μm)。使用cf4气体作为蚀刻气体而测定干蚀刻速度,求出实施例13-24和比较例7的干蚀刻速度比。干蚀刻速度比为(抗蚀剂下层膜)/(krf光致抗蚀剂)的干蚀刻速度比(表4)。
[0345]
[表4]
[0346]
表4干蚀刻速度比
[0347][0348]
如上所述,具有叔丁基或三氟甲基的酚醛清漆树脂可以通过变更分子骨架来调整为适应于工艺的蚀刻速度。
[0349]
(对高低差基板的被覆试验)
[0350]
作为对高低差基板的被覆试验,用200nm膜厚的sio2基板,进行了800nm沟槽区(trench)和未形成图案的开放区(open)的被覆膜厚的比较。将在实施例13-24和比较例7中调制的抗蚀剂下层膜形成用组合物涂布于上述基板后,在350℃烧成60秒,形成了约200nm的抗蚀剂下层膜。使用日立
ハイテクノロジーズ
株式会社制扫描型电子显微镜(s-4800)观察该基板的平坦化性,测定高低差基板的沟槽区(图案部)与开放区(无图案部)的膜厚差(沟槽区与开放区的涂布高低差,被称为偏差),从而评价了平坦化性。这里,所谓平坦化性,是指在图案存在的部分(trench(图案部))、与图案不存在的部分(开放区(无图案部)),在其上部存在的被涂布了的被覆物的膜厚差(iso-trench偏差)小(表5)。另外,将相对于比较例可以确认到小于10nm的改善的实施例评价为
△
,可以确认到10nm以上改善的实施例评价为
○
,相对于比较例可以确认到20nm以上改善的实施例评价为
◎
。
[0351]
[表5]
[0352]
表5平坦化性评价
[0353][0354]
如上所述,具有叔丁基或三氟甲基的酚醛清漆树脂显示良好的平坦化性。
[0355]
产业可利用性
[0356]
本发明涉及的酚醛清漆树脂不限于在低温烧成时,在高温烧成时也显示异常高的纯水接触角(=疏水性)。此外,本发明涉及的酚醛清漆树脂在混合交联剂、酸催化剂和表面活性剂而制成材料时,也在高温烧成时显示异常高的纯水接触角(=疏水性)。进一步,本发明涉及的酚醛清漆树脂显示良好的平坦化性,可以通过变更分子骨架来调整为适应于工艺的光学常数、蚀刻速度。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。