1.本发明涉及一种瓜蒌皮注射液核磁共振氢谱中独立信号峰的获取方法、定量分析方法、指纹图谱、应用。
背景技术:
2.瓜蒌皮注射液是以瓜蒌皮为原料,经水提醇沉,再经过离子交换树脂洗脱而制成的灭菌水溶液,具有扩张冠状动脉、保护心肌缺血的作用,临床上被广泛用于治疗冠心病和心绞痛。由于注射剂中的成分较为复杂,其质量评价和控制水平越发受人们重视。
3.申请公开号为cn100491998c的专利文献中公开了一种瓜蒌皮或瓜蒌皮注射液的液相色谱指纹图谱的测试方法,并构建瓜蒌皮注射液的液相指纹图谱作为产品质量控制的方法,但是该方法前处理复杂,需要对样品进行两次柱前衍生化,液相实验需要较长时间,洗脱程序用时长达60分钟,且该指纹图谱方法所关注的全是氨基酸类成分并没有考虑其他成分特别是潜在的起效成分。因此构建一种快速的可同时定量检测瓜蒌皮注射液的各种化学成分的方法对提升瓜蒌皮注射液质量控制有重大意义。
[0004]1h-nmr技术同时具备定量和定性的能力,具有前处理简单、快速、重现性好等优点。可以一次快速采集瓜蒌皮注射液中全部的成分信息用以定性和定量分析。相比于传统检测手段针对不同类别的物质需要用到不同方法,1h-nmr方法大大缩短了分析的时间,简化了操作,实现一种方法替代多种方法,单次分析替代多次分析。
技术实现要素:
[0005]
本发明解决的技术问题是为了克服现有技术中定量检测瓜蒌皮注射液成分的方法前处理复杂,检测时间长,仅可对单一类成分定量检测的缺陷,而提供了一种瓜蒌皮注射液核磁共振氢谱中独立信号峰的获取方法、定量分析方法、指纹图谱、应用。本发明中的定量分析方法可快速且可同时定量瓜蒌皮注射液32种化学成分,该方法所得到的指纹图谱可应用在瓜蒌皮注射液的质量评价与控制中。
[0006]
瓜蒌皮注射液成分复杂,因成分的h原子所处的化学环境相似,所以其1h-nmr谱图某些成分的信号峰存在严重的重叠问题,从而导致难以准确定量。本发明中,为解决此缺陷,经过大量的试验研究,发明人创造性地发现在信号峰重叠严重的光谱片段,先采用icoshift方法将谱图进行对齐然后通过mcr-als算法进行解卷积,最终通过浓度比例经过计算可得到待测成分的含量。此方法对定量检测瓜蒌皮注射液中化学成分的含量具有重要意义。此外,在定量检测瓜蒌皮注射液成分的过程中,发明人发现了5种未在瓜蒌皮注射液中报道过的成分,分别为:甘氨酸、鸟氨酸、蛋氨酸亚砜、胍基乙酸、葡萄糖。
[0007]
本发明通过以下技术方案解决上述问题。
[0008]
本发明提供了一种瓜蒌皮注射液1h-nmr图谱中独立信号峰的获取方法,其包括以下步骤:
[0009]
将所述瓜蒌皮注射液1h-nmr图谱中信号重叠处的信号进行信号处理获得独立信号峰,即可;
[0010]
所述信号重叠处中的信号重叠是指在同一光谱片段处,有两个或两个以上的成分具有信号峰,且通过线性拟合后无法将各个成分的信号峰分开;
[0011]
所述信号重叠处所对应的成分包括精氨酸、丝氨酸和d-哌啶酸;
[0012]
所述独立信号峰采用下述方法获得:
[0013]
1)采用icoshift不等间距分段对齐方法对所述信号重叠处的光谱片段进行对齐;
[0014]
当所述信号重叠处的信号所对应的成分为精氨酸时,选择3.2131-3.2825ppm光谱作为对齐片段,分为三段后进行对齐,第一分段为3.2131-3.2225ppm,第二分段为3.2225-3.2650ppm,第三分段为3.2650-3.2825ppm;
[0015]
当所述信号重叠处的信号所对应的成分为丝氨酸时,选择3.8099-3.8759ppm光谱作为对齐片段,分为两段后进行对齐,第一分段为3.8099-3.8440ppm,第二分段为3.8440-3.8759ppm;
[0016]
当所述信号重叠处的信号所对应的成分为d-哌啶酸时,选择2.9533-3.0041ppm光谱作为对齐片段,分为两段后进行对齐,第一分段为2.9533-2.9808ppm,第二分段为2.9808-3.0041ppm;
[0017]
2)采用mcr-als方法进行解卷积分离出独立信号峰。
[0018]
本发明中,所述瓜蒌皮注射液1h-nmr图谱可采用本领域常规的方法获得,例如采用下述方法获得:
[0019]
(1)供试样品溶液的制备:将所述瓜蒌皮注射液与含tsp的氘代水混合,配制供试样品溶液;
[0020]
(2)1h-nmr图谱的采集:将步骤(1)中所述的供试样品溶液置于核磁共振仪中进行测试,利用预饱和水峰压制脉冲序列对残余水峰进行压制,得到供试样品溶液的1h-nmr图谱,即可。
[0021]
步骤(1)中,所述瓜蒌皮注射液与所述含tsp(3-(三甲基甲硅烷基)丙酸-d4钠盐)的氘代水的体积比优选为(8-10):1,例如9:1。
[0022]
步骤(1)中,优选地,所述供试样品溶液的总体积为600~1000μl。
[0023]
步骤(1)中,在所述含tsp的氘代水中,所述tsp(内标)的浓度优选地为0.45-0.55mg/ml,例如0.506mg/ml。
[0024]
步骤(2)中,所述1h-nmr图谱的采集条件可为:探头温度为288-300k;脉冲序列noesygppr1d;谱宽为9.9974-12.9836ppm;中心频率为4.780-4.820ppm;弛豫延迟时间为13.0-25.0s;采集时间为2.27s;混合时间为50-100ms;采集次数为32或64次;增益值为32-57;检测数据点为32k或64k。
[0025]
步骤(2)中,所述1h-nmr图谱的采集条件还可为:探头温度为295-300k;脉冲序列noesygppr1d;以溶剂90%h2o 10%d2o锁场;谱宽为11.4912-12.4967ppm;中心频率为4.790-4.805ppm;弛豫延迟时间为14.5-15.5s;采集时间为2.27s;混合时间为50-100ms;采集次数为32次;增益值为32-57;检测数据点为32k或64k。
[0026]
步骤(2)中,所述1h-nmr图谱的采集条件还可为:探头温度为298k;脉冲序noesygppr1d;以溶剂90%h2o 10%d2o锁场;谱宽为12.016ppm;中心频率为4.800ppm;弛豫
延迟时间为15.0s;采集时间为2.27s;混合时间为50ms;采集次数为32次;增益值为44.3;检测数据点为64k。
[0027]
步骤(2)中,所述1h-nmr图谱进行数据采集时,优选地,需要进行调谐、探头匹配和匀场,并测定对应的90
°
脉冲宽度。
[0028]
本发明中,优选地,所述1h-nmr图谱进行所述信号处理前,还进行预处理。
[0029]
其中,所述的供试样品溶液的1h-nmr图谱的预处理可包括原始fid信号的傅里叶变换、基线校正、相位校正和图谱对齐。
[0030]
优选地,傅里叶变换之前,图谱以0.30hz的指数函数为窗函数进行处理。
[0031]
优选地,所述基线校正、相位校正和图谱对齐全部在mestrenova软件中自动进行。
[0032]
本发明中,所述mcr-als可为本领域常规的多元曲线分辨-交替最小二乘法。
[0033]
本发明还提供了一种瓜蒌皮注射液成分1h-nmr指纹图谱的构建方法,其包括以下步骤:将所述瓜蒌皮注射液的1h-nmr图谱进行信号归属,并指认化学成分的特征峰,即可;
[0034]
优选地,所述瓜蒌皮注射液的1h-nmr图谱采用如前所述的方法获得;
[0035]
优选地,所述瓜蒌皮注射液的1h-nmr图谱进行所述信号处理前,还进行如前所述的预处理。
[0036]
优选地,所述1h-nmr图谱分析需要用“分箱(binning)”方法进行导出。
[0037]
优选地,其中“分箱”方法包括但不限于“求和”、“平均值”、“分段积分”、“中位数”。优选地,所述分析的方法包括相似度评价方法和多变量统计方法;优选地,所述相似度评价方法包括皮尔森相关系数、夹角余弦值、欧氏距离和标准化欧氏距离;所述多变量统计方法包括主成分分析、层次聚类分析、偏最小二乘判别分析和正交偏最小二乘分析。
[0038]
本发明中,优选地,所述化学成分特征峰中包括15种氨基酸、7种小分子有机酸、4种核苷、4种生物碱及含氮化合物、1种糖和1种醇类化合物。
[0039]
本发明中,优选地,所述化学成分特征峰中包含32种化学成分,所述32种化学成分分别为:亮氨酸、异亮氨酸、缬氨酸、苏氨酸、丙氨酸、瓜氨酸、甘氨酸、脯氨酸、酪氨酸、苯丙氨酸、色氨酸、精氨酸、丝氨酸、鸟氨酸、蛋氨酸亚砜、γ-氨基丁酸、d-哌啶酸、胍基乙酸、烟酸、香草酸、甲酸、乙酸、胞苷、鸟苷、腺嘌呤、腺苷、水苏碱、葫芦巴碱、甜菜碱、3-羟基-2-甲基吡啶、葡萄糖、乙醇。
[0040]
本发明中,优选地,所述化学成分特征峰如表1所示。
[0041]
表1瓜蒌皮注射液归属的化学成分名称及特征峰信息
[0042]
[0043][0044]
本发明还提供一种采用所述瓜蒌皮注射液成分1h-nmr指纹图谱的构建方法得到的瓜蒌皮注射液成分1h-nmr指纹图谱。
[0045]
本发明还提供了一种瓜蒌皮注射液成分1h-nmr指纹图谱,其特征成分如下表所示:
[0046]
[0047]
。
[0048]
本发明还提供了一种瓜蒌皮注射液成分的定量分析方法,其包括以下步骤:
[0049]
①
:将所述瓜蒌皮注射液的1h-nmr图谱进行信号归属获得独立信号峰;
②
:通过所述独立信号峰对归属出的化学成分进行含量测定,即可;
[0050]
s1.对于信号峰无重叠的化学成分所对应的特征峰,采用直接积分或利用线性拟合方法积分获得其对应成分含量;
[0051]
s2.对于信号峰重叠的化学成分所对应的特征峰,采用所述瓜蒌皮注射液1h-nmr图谱中独立信号峰的获取方法分离出的独立信号峰,根据所述信号峰重叠的化学成分的标准品的浓度与所述瓜蒌皮注射液中化学成分的浓度比例,计算得到所述信号峰重叠的化学成分在所述瓜蒌皮注射液中含量。
[0052]
本发明中,优选地,所述瓜蒌皮注射液成分的定量分析方法中,用于含量测定的特征峰如表2所示。
[0053]
表2瓜蒌皮注射液中需经直接积分、线性拟合或mcr-als处理的化学成分名称及特征峰信息
[0054]
[0055][0056]
本发明中,所述含量测定方法优选地为以tsp为内标定量,按式(1)计算。
[0057][0058]
其中,c
x
和cr分别是待测化学成分和内标的质量浓度;s
x
和sr分别是待测化学成分和内标的特征峰面积;n
x
和nr分别是待测化学成分和外标的特征峰质子数;m
x
和mr分别是待测化学成分和外标的相对分子量。
[0059]
优选地,步骤s2中所述瓜蒌皮注射液1h-nmr图谱中独立信号峰的获取方法中mcr-als基于双线性模型将光谱矩阵d被分解成浓度矩阵c和纯光谱矩阵s
t
的乘积,这个模型可以表示为式(2),浓度矩阵c表示的是各光谱中待测成分的浓度。
[0060]
d=cs
t
e(2)
[0061]
其中,e是误差矩阵。
[0062]
优选地,瓜蒌皮注射液样品中的化学成分有32个,分别为亮氨酸、异亮氨酸、缬氨酸、苏氨酸、丙氨酸、瓜氨酸、甘氨酸、脯氨酸、酪氨酸、苯丙氨酸、色氨酸、精氨酸、丝氨酸、鸟氨酸、蛋氨酸亚砜、γ-氨基丁酸、胍基乙酸、d-哌啶酸、烟酸、香草酸、甲酸、乙酸、胞苷、鸟苷、腺嘌呤、腺苷、水苏碱、葫芦巴碱、甜菜碱、3-羟基-2-甲基吡啶、葡萄糖、乙醇。
[0063]
本发明中,所述瓜蒌皮注射液可以为上海上药第一生化药业有限公司所生产的瓜蒌皮注射液,例如批号为:2103212的瓜蒌皮注射液。
[0064]
本发明还提供一种如前所述的经定量检测瓜蒌皮注射液中32种化学成分的方法得到的指纹图谱。
[0065]
本发明还提供一种如前所述的定量检测瓜蒌皮注射液中成分的方法和/或所述指纹图谱在瓜蒌皮注射液质量评价及控制中的应用。
[0066]
本发明所用试剂和原料均市售可得。
[0067]
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0068]
和现有检测方法相比,本发明的有益效果在于:
[0069]
本发明提供的含量测定方法基于1h-nmr技术,操作简单、快速,相较于高效液相色谱法,预处理简单,不破坏样品,检测完成后样品可回收,具体如下:
[0070]
(1)检测速度快,一次测定只需15分钟,大大缩短了检测时间。
[0071]
(2)可以同时且单次测定各个化学成分的含量。本发明nmr定量可以测定单个氨基酸的含量各个化学成分的含量,更进一步地,可以针对性的得到单个氨基酸的含量变化更
有利于生产中的质量控制和质量评价。
[0072]
(3)可以解决因信号重叠而无法通过积分计算成分含量的问题,更大程度上挖掘和利用nmr光谱中的成分信息,在不依赖其他分析手段的前提下,对注射液中更多的成分进行含量计算。
[0073]
(4)定量时无需每种化合物的对照品,无需绘制标准曲线,只需要内标即可定量。
[0074]
(5)本发明提供的基于1h-nmr技术的方法具有良好的重现性。
[0075]
(6)本发明还发现了瓜蒌皮注射液中未报道过的5种化学成分,分别是:甘氨酸、鸟氨酸、蛋氨酸亚砜、胍基乙酸、葡萄糖。
附图说明
[0076]
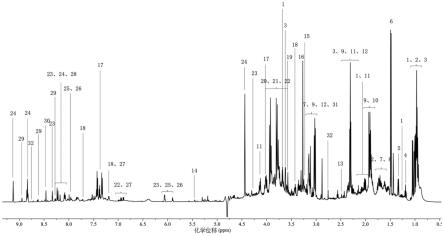
图1为瓜蒌皮注射液的代表性1h-nmr指纹图谱及其信号峰归属。
[0077]
图2为为瓜蒌皮注射液及注射液加甘氨酸标准品的1h-nmr图谱。
[0078]
图3为瓜蒌皮注射液及注射液加鸟氨酸标准品的1h-nmr图谱。
[0079]
图4为瓜蒌皮注射液及注射液加蛋氨酸亚砜标准品的1h-nmr图谱。
[0080]
图5为瓜蒌皮注射液及注射液加胍基乙酸标准品的1h-nmr图谱。
[0081]
图6为瓜蒌皮注射液及注射液加葡萄糖标准品的1h-nmr图谱。
[0082]
图7为利用icoshift对精氨酸图谱片段分三段不等间距峰对齐后得到的结果。
[0083]
图8为利用matlab软件进行mcr-als处理后得到的精氨酸结果示意图。
[0084]
图9为利用icoshift对精氨酸图谱片段等间距分五段峰对齐后得到的结果。
[0085]
图10为利用icoshift对丝氨酸图谱片段分两段不等间距峰对齐后得到的结果。
[0086]
图11为利用matlab软件进行mcr-als处理后得到的丝氨酸结果示意图。
[0087]
图12为利用icoshift对d-哌啶酸图谱片段分两段不等间距峰对齐后得到的结果。
[0088]
图13为利用matlab软件进行mcr-als处理后得到的d-哌啶酸结果示意图。
具体实施方式
[0089]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0090]
以下实施例和对比例中,使用的核磁共振光谱仪为bruker advanced iii 600型核磁共振光谱仪。
[0091]
实施例1
[0092]
一种定量检测瓜蒌皮注射液成分的方法,包括如下步骤:
[0093]
(1)含tsp的氘代水配制:精确称取10.12mg的tsp于20ml容量瓶,加入氘代水定容至刻度,充分溶解并混合均匀。
[0094]
(2)瓜蒌皮注射液供试样品的配制:精确移取瓜蒌皮注射液540μl和含tsp的氘代水60μl于2ml离心管,混合均匀,制成瓜蒌皮注射液供试样品。
[0095]
(3)1h-nmr图谱的采集:将供试样品置于核磁共振仪中进行测试,利用预饱和水峰压制脉冲序列对残余水峰进行压制,得到供试样品的1h-nmr图谱。1h-nmr采集仪器是bruker advanced iii 600型核磁共振光谱仪(bruker,德国,配24位自动进样器与5mm bbo探头,
topspin工作站)。1h-nmr数据采集时需要进行调谐、探头匹配和匀场,并测定对应的90
°
脉冲宽度。
[0096]1h-nmr采集参数为:探头温度为298k;脉冲序noesygppr1d;以溶剂90%h2o 10%d2o锁场;谱宽为12.016ppm;中心频率为4.800ppm;弛豫延迟时间为15.0s;采集时间为2.27s;混合时间为50ms;采集次数为32次;增益值为44.3;检测数据点为64k。
[0097]
(4)1h-nmr原始数据预处理:采集到的原始图谱应用以0.30hz的指数函数为窗函数进行处理,并进行fid信号的傅里叶变换,而后将图谱导入mestrenova软件中以tsp(0.0)为化学位移参考定标,自动进行基线和相位校正。
[0098]
(5)瓜蒌皮注射液1h-nmr图谱的归属与指纹图谱的构建:瓜蒌皮注射液1h-nmr图谱结构见图1,图1中各编号所指代的化合物详见表3,其中甘氨酸、鸟氨酸、蛋氨酸亚砜、胍基乙酸、葡萄糖这5种化学成分,为在瓜蒌皮注射液中未报道过的成分,其谱图如图2-图6所示。
[0099]
表3瓜蒌皮注射液归属的化学成分名称及特征峰信息
[0100]
[0101][0102]
[0103]
(6)特征峰的积分与化学成分的定量:瓜蒌皮注射液中共有32个化学成分高于定量限,分别为:亮氨酸、异亮氨酸、缬氨酸、苏氨酸、丙氨酸、瓜氨酸、甘氨酸、脯氨酸、酪氨酸、苯丙氨酸、色氨酸、精氨酸、丝氨酸、鸟氨酸、蛋氨酸亚砜、γ-氨基丁酸、胍基乙酸、d-哌啶酸、烟酸、香草酸、甲酸、乙酸、胞苷、鸟苷、腺嘌呤、腺苷、水苏碱、葫芦巴碱、甜菜碱、3-羟基-2-甲基吡啶、葡萄糖、乙醇。用mestrenova软件中线性拟合的方法对除信号重叠严重精氨酸、丝氨酸、d-哌啶酸外的其他29个成分的特征峰以及tsp的内标峰进行积分。而后,根据式(1)计算上述成分的浓度,结果如表4所示。
[0104]
表4瓜蒌皮注射液中成分浓度结果
[0105][0106]
实施例2
[0107]
瓜蒌皮注射液的1h-nmr定量方法学考察:
[0108]
(1)线性关系:
①
用去离子水和含内标tsp的氘代水以9:1的比例配制氘代溶剂。
②
取安瓿瓶分别加入0.2、0.4、0.6、0.8、1.2、1.4ml瓜蒌皮注射液,冻干后加入600μl氘代溶剂复溶即得供试品样品。
③
用实施例1中步骤(3)所述方法采集所有样品的1h-nmr图谱,并用实施例1中步骤(6)所述方法对各成分特征峰积分。
④
以对照品的特征峰面积为纵坐标,质量浓度为横坐标进行线性回归,得到回归方程的系数r2均大于0.999,证明nmr仪器的线性关系良好。
[0109]
(2)专属性:取瓜蒌皮注射液分别按实施例1和2中步骤(1)所述方法配制供试液样品,通过hsqc法验证定量峰的专属性,结果表明定量峰均无杂质峰干扰,说明该方法专属性良好。
[0110]
(3)仪器精密度:取瓜蒌皮注射液按实施例1中步骤(1)所述方法配制供试液样品,用步骤(3)所述方法连续采集六次1h-nmr图谱,并用步骤(6)所述方法对各特征峰积分并计算各化学成分和外标的峰面积比值及rsd值,结果表明各特征峰的rsd值均小于2.92%,证
明该方法仪器精密度良好。
[0111]
(4)制样重复性:取瓜蒌皮注射液分别按实施例1中步骤(1)所述方法平行配制6份供试液样品,用实施例1中步骤(3)所述方法分别采集六个平行样品的1h-nmr图谱,并用实施例1和2中步骤(6)所述方法对各特征峰积分并计算各化学成分和外标的峰面积比值及rsd值,结果表明各特征峰的rsd值均小于2.97%,证明该样品制备方法重复性良好。
[0112]
(5)9h稳定性:取瓜蒌皮注射液分别按实施例1中步骤(1)所述方法配制供试液样品,用实施例1中步骤(3)所述方法在第0h,3h,6h,9h,采集1h-nmr图谱,并用实施例1中步骤(6)所述方法对各特征峰积分并计算各化学成分和外标的峰面积比值及rsd值,结果表明9h内未出现新的特征峰,且各特征峰的rsd值均小于2.95%,证明该供试液样品在9h内稳定性良好。
[0113]
(6)加样回收实验:取已知含量的瓜蒌皮注射液,分高、中、低三个浓度组加入定量的对照品而后用已加入对照品的瓜蒌皮注射液按实施例1中步骤(1)所述方法配制供试液样品,并用实施例1中步骤(3)-(6)所述方法进行含量测定,并计算各化学成分的回收率。结果表明各成分的回收率为97.39%~102.94%,说明该方法对瓜蒌皮注射液定量准确度良好。
[0114]
实施例3
[0115]
基于1h-nmr结合icoshift算法及mcr-als计算精氨酸含量,包括如下步骤:
[0116]
(1)精氨酸母液配制:精确称取9.54mg的精氨酸于5ml容量瓶,加入去离子水定容至刻度,充分溶解并混合均匀。
[0117]
(2)含不同浓度精氨酸的瓜蒌皮注射液样品配制:取安瓿瓶分别加入0、0.1、0.15、0.2、0.25、0.3ml精氨酸母液,冻干后分别加入瓜蒌皮注射液540μl和含tsp的氘代水60μl于2ml离心管,混合均匀,按顺序即得注射液光谱1和注射液加样光谱2-6。另外取0.3ml精氨酸母液,用去离子水和含内标tsp的氘代水配制成总体积为600μl含10%氘代的纯精氨酸样品。
[0118]
(3)1h-nmr图谱采集:用实施例1中步骤(3)所述方法采集所有样品的1h-nmr图谱。
[0119]
(4)谱图处理:用实施例1中步骤(3)所述方法处理采集所有样品的1h-nmr图谱。选取化学位移为3.2131-3.2825的部分导出csv文件,之后利用icoshift算法在matlab中进行不等间距分三段的峰对齐,对齐后的谱图见图7。
[0120]
不等间距分三段的峰如下:选择3.2131-3.2825ppm光谱作为对齐片段,分为三段后进行对齐,第一分段为3.2131-3.2225ppm,第二分段为3.2225-3.2650ppm,第三分段为3.2650-3.2825ppm。
[0121]
(5)精氨酸含量计算:在进行mcr-als计算之前需要确定光谱中的组分数。本实施例的目的在于解出精氨酸的浓度,此将精氨酸视为一个组分,剩下的混合物视为另外一个组分。利用mcr-als工具箱对对齐后的谱图进行解卷积得到的谱图如图8,其中纯精氨酸和注射液中精氨酸的浓度比例为1:0.4467,根据纯精氨酸谱的浓度计算得到瓜蒌皮注射液中的精氨酸浓度为0.426mg/ml,具体浓度输出数据如表5。
[0122]
表5精氨酸光谱片段输出浓度矩阵结果
[0123][0124]
(6)mcr-als结果验证:为了验证mcr-als结果的准确性,以实际加样量为y,计算得到的加样量为x绘制回归曲线得到r2=0.996;另外还计算了mcr-als解出的精氨酸光谱和精氨酸实验光谱之间的相关系数,结果为0.981;这说明该方法可以较为准确的从混合物光谱中提取精氨酸纯光谱的信息。
[0125]
实施例4
[0126]
基于1h-nmr结合icoshift算法及mcr-als计算丝氨酸含量,包括如下步骤:
[0127]
(1)丝氨酸母液配制:精确称取15.75mg的丝氨酸于5ml容量瓶,加入去离子水定容至刻度,充分溶解并混合均匀。
[0128]
(2)含不同浓度丝氨酸的瓜蒌皮注射液样品配制:取安瓿瓶分别加入0、0.025、0.05、0.1、0.15、0.2ml丝氨酸母液,用实施例3步骤(2)所述方法制备注射液光谱1和注射液加样光谱2-6。另外取0.2ml丝氨酸母液,用实施例3步骤(2)所述方法制备纯丝氨酸样品。
[0129]
(3)1h-nmr图谱采集:用实施例1中步骤(3)所述方法采集所有样品的1h-nmr图谱。
[0130]
(4)谱图处理:用实施例1中步骤(3)所述方法处理采集所有样品的1h-nmr图谱。选取化学位移为3.8099-3.8759的部分导出csv文件,之后利用icoshift算法在matlab中进行不等间距分两段的峰对齐,对齐后的谱图见图10。
[0131]
不等间距分两段的峰如下:选择3.8099-3.8759ppm光谱作为对齐片段,分为两段后进行对齐,第一分段为3.8099-3.8440ppm,第二分段为3.8440-3.8759ppm。
[0132]
(5)丝氨酸含量计算:用实施例3步骤(5)所述方法对丝氨酸浓度进行计算得到瓜蒌皮注射液中的丝氨酸浓度为0.332mg/ml,具体浓度输出数据如表6,具体结果见图11。
[0133]
(6)mcr-als结果验证:用实施例3步骤(6)所述方法验证mcr-als结果的准确性,绘制回归曲线得到r2=0.999;mcr-als解出的丝氨酸光谱和丝氨酸实验光谱之间的相关系数,结果为0.999。
[0134]
表6丝氨酸光谱片段输出浓度矩阵结果
[0135][0136]
实施例5
[0137]
基于1h-nmr结合icoshift算法及mcr-als计算d-哌啶酸含量,包括如下步骤:
[0138]
(1)d-哌啶酸母液配制:精确称取5.20mg的d-哌啶酸于5ml容量瓶,加入去离子水定容至刻度,充分溶解并混合均匀。
[0139]
(2)含不同浓度d-哌啶酸的瓜蒌皮注射液样品配制:取安瓿瓶分别加入0、0.025、0.05、0.1、0.15、0.2mld-哌啶酸母液,用实施例3步骤(2)所述方法制备注射液光谱1和注射液加样光谱2-6。另外取0.3ml d-哌啶酸母液,用实施例3步骤(2)所述方法制备纯d-哌啶酸样品。
[0140]
(3)1h-nmr图谱采集:用实施例1中步骤(3)所述方法采集所有样品的1h-nmr图谱。
[0141]
(4)谱图处理:用实施例1中步骤(3)所述方法处理采集所有样品的1h-nmr图谱。选取化学位移为2.9533-3.0041的部分导出csv文件,之后利用icoshift算法在matlab中进行不等间距分两段的峰对齐,对齐后的谱图见图12。
[0142]
不等间距分两段的峰如下:选择2.9533-3.0041ppm光谱作为对齐片段,分为两段后进行对齐,第一分段为2.9533-2.9808ppm,第二分段为2.9808-3.0041ppm。
[0143]
(5)d-哌啶酸含量计算:用实施例3步骤(5)所述方法对d-哌啶酸浓度进行计算得到瓜蒌皮注射液中的d-哌啶酸浓度为0.542mg/ml,具体浓度输出数据如表7,具体结果见图13。
[0144]
(6)mcr-als结果验证:用实施例3步骤(6)所述方法验证mcr-als结果的准确性,绘制回归曲线得到r2=0.999;mcr-als解出的d-哌啶酸光谱和d-哌啶酸实验光谱之间的相关系数,结果为0.992。
[0145]
表7d-哌啶酸光谱片段输出浓度矩阵结果
[0146][0147]
对比例1
[0148]
对比例1与实施例3的区别仅在于:未采取实施例3步骤(4)中的icoshift算法对齐操作。由此得到的精氨酸的浓度结果如表8所示。
[0149]
表8未对齐光谱进行mcr-als后浓度矩阵输出结果
[0150][0151]
由上表发现,注射液加样光谱2-6,随着精氨酸母液的加量越多,但在加样光谱4和5中通过mcr-als解出的精氨酸的浓度出现减小的趋势,说明经过未进行对齐操作直接进行mcr-als解出的浓度出现错误,即对于重叠严重的峰型来说,进行icoshift对齐操作对mcr-als解出精确的结果有着重要意义同时也是必不可少的。
[0152]
对比例2
[0153]
对比例2与实施例3的区别仅在于:步骤(4)中利用icoshift算法在matlab中进行等间距分五段的峰对齐,对齐后的谱图见图10。由此得到的精氨酸的浓度结果如表9所示,以实际加样量为y,计算得到的加样量为x绘制回归曲线得到r2=0.995;但是计算mcr-als解出的精氨酸光谱和精氨酸实验光谱之间的相关系数,得到结果为0.776,这说明相较于实施例3,等间距分五段后经过mcr-als解出的结果与实际浓度之间的线性良好,但是解得的纯物质光谱和实际光谱间存在较大差异,二者相关性较低,无法解出准确的纯物质光谱。在
使用icoshift进行对齐时,多数是通过等间距分段,尽量让每个峰都在一段后分段对齐,如图9,是对精氨酸光谱监督等间距分五段对齐的结果,很明显可以发现在四、五两段分段处出现了峰变形的情况,这是最后输出谱图和实验谱图相关性较低的成因。因此等间距分五段对齐对于精氨酸的光谱片段不适用。
[0154]
表9等间距五段光谱对齐后进行mcr-als后浓度矩阵输出结果
[0155][0156][0157]
上述详细说明是针对本发明其中之一可行实施例的具体说明,该实施例并非用以限制本发明的专利范围,凡未脱离本发明所为的等效实施或变更,均应包含于本发明技术方案的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。