1.本发明属于药物分析技术领域,具体涉及一种右旋雷贝拉唑钠原料药有关物质的检测方法,该检测方法采用高效液相色谱法对右旋雷贝拉唑钠及13种有关杂质进行定性或定量分析。
背景技术:
2.右旋雷贝拉唑是一种质子泵抑制剂(ppi),它通过与胃壁细胞分泌表面的(h
,k
)-atp酶系统的2个位点产生共价结合,抑制胃酸生成的最后一步。右雷贝拉唑钠类似于雷贝拉唑钠,是一种前药,在胃壁细胞处质子化,形成活性的亚磺酰胺。用于胃、十二指肠溃疡以及胃食管返流症(gerd)的短期治疗。
3.有关物质是在药物合成生产过程中带入的起始物料、中间体、副反应产物和降解杂质等,对有关物质进行检测可以对药物的质量和安全性进行控制。目前的国内外药典尚未收录右旋雷贝拉唑钠有关物质的检测方法,对雷贝拉唑钠的有关物质检测方法在中国药典、ep、usp及jp药典中均有收录,其中中国药典和jp药典洗脱条件采用等度洗脱方式,对于极性较小的杂质,较难被洗脱出来,从而影响分析结果准确性;usp药典流动相洗脱方式较繁琐,流动相双项双混合,且研究杂质个数较少(4个),不能满足检测需要;ep药典中流动相为三相梯度洗脱,而梯度洗脱在三相混合时,容易产生气泡影响基线平稳,干扰某些杂质的有效检出,同时三相混合对仪器精密度要求较高,在流动相洗脱比例微小变动时容易产生较大误差。
4.中国专利cn112834628a公开一种高效液相色谱法测定雷贝拉唑钠及其杂质的方法,以磷酸盐缓冲液和乙腈为流动相进行梯度洗脱检测包含雷贝拉唑钠和9种常见杂质,分析杂质的种类较少,灵敏度和准确度偏低,仍无法满足右旋雷贝拉唑钠原料药生产过程中的质量监测的要求。
5.为了保证药物的安全有效,需要对药物中的有关物质进行研究、检测和监控。有关物质主要为工艺副产物及降解产物,药品在放置过程中,杂质谱在发生变化,因此,需要根据不同的合成路线和生产工艺、贮藏条件建立合适的分析方法,达到对右旋雷贝拉唑钠原料药有关物质准确、有效的检测和监控。
技术实现要素:
6.本发明的目的是在现有技术的基础上,提供一种右旋雷贝拉唑钠原料药有关物质的检测方法,可以在一次色谱行为中检测13种相关杂质,便于生产过程中产品质量的控制,简单易行,准确度及精密度高,重现性良好。
7.本发明的技术方案如下:
8.一种右旋雷贝拉唑钠原料药中有关物质的检测方法,该检测方法采用高效液相色谱对右旋雷贝拉唑钠及有关物质进行定性或定量检测,其高效液相色谱条件包括:色谱柱为岛津wondasil c
18-wr柱;采用流动相a和流动相b为混合流动相进行梯度洗脱,流动相a为
15~25mmol/l磷酸二氢钾缓冲溶液,调节其ph至6.0~8.0;流动相b为甲醇。
9.在梯度洗脱过程中流动相a和流动相b的初始比例为73~77:27~23;在0-8分钟内,流动相a和流动相b的体积比保持初始比例不变;在8-18分钟内,流动相a和流动相b的体积比由初始比例匀速渐变至55:45;在18-33分钟内,流动相a和流动相b的体积比保持55:45不变;在33-75分钟内,流动相a和流动相b的体积比由55:45匀速渐变至25:75;在75-95分钟内,流动相a和流动相b的体积比保持25:75不变;在95-96分钟内,流动相a和流动相b的体积比由25:75匀速渐变至初始比例;在96-106分钟内,流动相a和流动相b的体积比保持初始比例不变。
10.对于本发明而言,在梯度洗脱过程中,控制流动相a和流动相b的初始比例为73~77:27~23,可以但不局限于73:27、74:26、75:25、76:24或77:23,在一种优选方案中,流动相a和流动相b的初始比例为75:25。
11.在一种优选方案中,当流动相a和流动相b的初始比例为75:25;所述梯度洗脱包括以下步骤:在0-8分钟内,流动相a和流动相b的体积比保持75:25不变;在8-18分钟内,流动相a和流动相b的体积比由初始比例匀速渐变至55:45;在18-33分钟内,流动相a和流动相b的体积比保持55:45不变;在33-75分钟内,流动相a和流动相b的体积比由55:45匀速渐变至25:75;在75-95分钟内,流动相a和流动相b的体积比保持25:75不变;在95-96分钟内,流动相a和流动相b的体积比由25:75匀速渐变至75:25;在96-106分钟内,流动相a和流动相b的体积比保持75:25不变。具体的梯度洗脱过程如下表1:
12.表1梯度洗脱过程
[0013][0014][0015]
本发明采用高效液相色谱法检测时,流动相a为15~25mmol/l磷酸二氢钾缓冲溶液,调节其ph至6.0~8.0;流动相b为甲醇。在一种优选方案中,流动相a为20mmol/l磷酸二氢钾缓冲溶液,调节其ph至6.8~7.2。特别优选地,流动相a为20mmol/l磷酸二氢钾缓冲溶液,调节其ph至7.0。
[0016]
在色谱法中,色谱柱的选择十分重要,对色谱柱的要求:柱效高、选择性好,分析速度快等。本发明采用高效液相色谱对右旋雷贝拉唑钠及有关物质进行定性或定量检测时,色谱柱为岛津wondasil c
18-wr柱,在色谱分析的过程中监测的杂质种类及个数多,各杂质间及杂质与主成分间分离度良好,能快速准确的监控右旋雷贝拉唑钠中的有关物质。在不影响检测效果的情况下,优选色谱柱的长度为250mm,直径为4.6mm,填料粒径为5μm,即本发明采用的色谱柱为岛津wondasil c
18-wr柱(250x4.6mm,5μm)。在实验过程中发现,采用其他类似的色谱柱,例如,inertsil ods-3,在色谱分析的过程中,部分杂质与主成分的分离度
不佳,甚至无法达到基线分离。
[0017]
对于本发明而言,需要检测13种不同的杂质,包括中间体杂质、工艺杂质和降解杂质,杂质的种类较多,在色谱分析的过程中,需要严格控制色谱分析过程中的柱温,当色谱分析选择的柱温偏高或偏低时,容易导致某些杂质之间的分离度低于1.5,分离度不佳,不容易达到基线分离。在本发明中,控制柱温为18~22℃,可以但不局限于18℃、19℃、20℃、21℃或22℃,优选地,控制柱温为20℃。
[0018]
进一步的,高效液相色谱条件包括:检测波长为200~400nm,优选280nm。
[0019]
进一步地,流速为0.5~1.5ml/min;优选为1.0ml/min。
[0020]
进一步地,进样量为5~20μl,可以但不局限于5μl、10μl、15μl或20μl;优选地,进样量为10μl。
[0021]
本发明提及的右旋雷贝拉唑钠原料药中有关物质,包括以下物质:杂质1:2-氯甲基-4-(3-甲氧基丙氧基)-3-甲基吡啶;杂质2:2-巯基苯并咪唑;杂质3:2-[[4-(3-甲氧基丙氧基)-3-甲基吡啶-2-基]-甲硫基]-1h-苯并咪唑;杂质4:2-[[[4-(3-甲氧基丙氧基)-3-甲基-2-吡啶基]甲基]砜]-1h-苯并咪唑钠盐;杂质5:2-氨基苯并咪唑;杂质6:2-[[[4-(3-甲氧基丙氧基)-3-甲基-2-吡啶基]甲基]亚砜]-1h-苯并咪唑氮氧化物;杂质7:2-羟基苯并咪唑;杂质8:2-[[[4-(3-甲氧基丙氧基)-3-甲基-2-吡啶基]甲基]砜]-1h-苯并咪唑氮氧化物;杂质9:苯并咪唑-2-磺酸;杂质10:1-(1h-苯并咪唑-2-基)-1,4-二羟基-3-甲基-4-氧化-2-吡啶甲酸钠;杂质20:2-[[(4-氯-3-甲基-2-吡啶基)甲基]亚磺酰基]-1h-苯并咪唑;杂质21:2-[[(4-甲氧基-3-甲基-2-吡啶基)甲基]亚磺酰基]-1h-苯并咪唑;杂质22:2-[(4-甲氧基-3-甲基吡啶-2-基)甲硫基]-1h-苯并咪唑。
[0022]
上述提及的有关物质的结构式如下:
[0023]
[0024][0025]
本发明采用高效液相色谱法,分别从色谱条件等方面进行筛选、优化,拟定有关物质方法,进行方法学验证,对杂质1(sma)进行定性研究、对杂质2(smb)、杂质3(中间体1)、杂质4~10,20~22(工艺杂质及降解杂质)进行了定量研究,并提供完整的验证方案,操作简单,稳定性和重现性良好。其中,杂质2的校正因子为0.57,杂质3的校正因子为0.88,杂质4的校正因子为1.13,杂质5的校正因子为0.71,杂质6的校正因子0.67,杂质7的校正因子为0.74,杂质8的校正因子为0.88,杂质9的校正因子为0.93,杂质10的校正因子为0.69,杂质20的校正因子为0.76,杂质21的校正因子为0.81,杂质22的校正因子为0.81。
[0026]
在一种优选方案中,本发明提供的右旋雷贝拉唑钠原料药中有关物质的检测方法,包括如下操作步骤:
[0027]
(1)溶液配制:
[0028]
取右旋雷贝拉唑钠样品适量,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】溶解并定量稀释制成每1ml约含右旋雷贝拉唑钠1.0mg的溶液,作为供试品溶液。精密量取供试品溶液1.0ml,置100ml量瓶中,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释至刻度,摇匀,作为对照溶液。
[0029]
(2)将供试品溶液和杂质对照品溶液分别注入液相色谱仪,记录色谱图,按外标法以峰面积计算,色谱条件如下:
[0030]
色谱柱:岛津wondasil c
18-wr柱(250x4.6mm,5μm);
[0031]
柱温为:18~22℃,优选为20℃;
[0032]
检测波长:200-400nm,进一步,波长为280nm;
[0033]
流速为:0.5~1.5ml/min;优选为1.0ml/min;
[0034]
进样量:5~20μl;优选10μl;
[0035]
流动相a:20mmol/l磷酸二氢钾缓冲溶液,调节其ph为6.0-8.0;优选地,调节其ph
羟基苯并咪唑(杂质7,trc)、2-[[[4-(3-甲氧基丙氧基)-3-甲基-2-吡啶基]甲基]砜]-1h-苯并咪唑氮氧化物(杂质8,std)、苯并咪唑-2-磺酸(杂质9,std)、1-(1h-苯并咪唑-2-基)-1,4-二羟基-3-甲基-4-氧化-2-吡啶甲酸钠(杂质10,usp)、2-[[(4-氯-3-甲基-2-吡啶基)甲基]亚磺酰基]-1h-苯并咪唑(杂质20,std)、2-[[(4-甲氧基-3-甲基-2-吡啶基)甲基]亚磺酰基]-1h-苯并咪唑(杂质21,std)、2-[(4-甲氧基-3-甲基吡啶-2-基)甲硫基]-1h-苯并咪唑(杂质22,std)、甲醇(色谱纯,上海星可高纯溶剂有限公司)、磷酸二氢钾(分析纯,国药集团化学试剂有限公司)、氢氧化钠(分析纯,上海泰坦科技股份有限公司)、超纯水(自制,millipore)。
[0050]
2.仪器:具体仪器的名称和规格见下表2。
[0051]
表2具体仪器的名称和规格
[0052]
fa124万分之一天平上海舜宇恒平科学仪器有限公司xpe204万分之一天平梅特勒auw 120d十万分之一天平日本岛津sqp十万分之一天平赛多利斯科学仪器有限公司xp6i038百万分之一天平梅特勒phs-3c数字酸度计上海仪电科学仪器股份有限公司agilent 1100高效液相色谱仪安捷伦
[0053]
二、液相色谱条件
[0054]
色谱柱采用岛津wondasil c
18-wr柱(250x4.6mm,5μm);以20mmol/l磷酸二氢钾缓冲溶液(采用氢氧化钠调节其ph至7.0)为流动相a,以甲醇为流动相b,按下表1进行梯度洗脱;流速为1.0ml/min;柱温20℃;检测波长为280nm,精密量取供试品溶液10μl,注入液相色谱仪,记录色谱图。
[0055][0056]
三、实验过程
[0057]
1.右旋雷贝拉唑钠原料药中有关物质的检测
[0058]
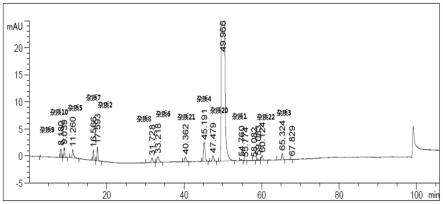
取本品(批号为:180923421)适量,精密称定,用溶剂[0.01mol/l氢氧化钠-甲醇(40:60)]溶解并定量稀释制成每1ml约含右旋雷贝拉唑钠1mg的溶液,作为供试品溶液。精密量取供试品溶液1.0ml,置100ml量瓶中,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释至刻度,摇匀,作为对照溶液。量取对照溶液10μl注入液相色谱仪,按照上述液相色谱条件调节检测灵敏度,使主成分色谱峰的峰高约为满量程的15%~20%;再精密量取供试品溶液、对照溶液各10μl,分别注入液相色谱仪,记录色谱图,如图1所示。采用加校正
因子的自身对照对已知杂质进行定量,采用自身对照法对未知杂质进行测定。检测结果如表3所示。按照上述测试方法,将样品(批号为:181024421以及181105421)进行检测,结果见表3。
[0059]
表3不同批次供试品测试结果
[0060][0061]
2.方法学验证
[0062]
2.1专属性
[0063]
取右旋雷贝拉唑钠样品(批号为:180923421)适量(约10mg),精密称定,置10ml量瓶中,用溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】溶解并稀释至刻度,摇匀,作为供试品溶液。取杂质1、2、3、4、5、6、7、8、9、10、20、21、22各适量,分别用溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】溶解并制成每1ml中各约含100μg的溶液,作为杂质母液;另称取右旋雷贝拉唑钠样品约10mg置10ml量瓶中,加入上述杂质母液适量,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释至刻度,使得杂质2、3、5、6、7、8、9、10、20、21和22为1μg/ml(其中,杂质1为10μg/ml,杂质4为5μg/ml,主成分的浓度为1mg/ml),摇匀,作为杂质与样品混合溶液。
[0064]
称取右旋雷贝拉唑钠样品约10mg,置10ml量瓶中,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】溶解并稀释至刻度,作为供试品溶液。
[0065]
分别取各杂质母液适量,用溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释配制成与一定浓度的溶液作为单个杂质定位溶液。
[0066]
精密量取溶剂、杂质与样品混合溶液、供试品溶液、单个杂质定位溶液各10μl,注入液相色谱仪,记录色谱图,结果见表4,相关谱图见图1-图2。
[0067]
表4专属性试验结果
[0068]
杂质与样品混合溶液保留时间min理论板数分离度杂质98.1808149/杂质109.03965872.12杂质511.26064524.41杂质716.5663079711.29杂质217.593354372.73杂质831.7282674324.55杂质633.218209601.76杂质2140.362356248.05杂质445.191611836.08杂质2047.479664973.11主成分49.966865233.51杂质154.760993716.97
未知杂质155.7741166081.50未知杂质258.0821535393.70未知杂质359.0831672871.71杂质2260.1241362771.69杂质365.3241929718.33未知杂质467.8292120864.23
[0069]
结果表明:该色谱条件下,基线平稳,空白溶剂对本品测定无干扰,右旋雷贝拉唑钠主峰理论塔板数为88655,主峰与相邻杂质分离良好。杂质样品混合溶液中,各物质峰形较好,主峰与相邻杂质的分离度均为1.5以上,各杂质间的最小分离度为1.50,表明专属性良好。
[0070]
2.2破坏试验
[0071]
为考察在所选择的色谱条件下能否检出右旋雷贝拉唑钠可能产生的降解产物,分别用高温、酸、碱、氧化、光照等剧烈条件对本品进行破坏,将破坏后的样品用溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】溶解制备成供试品溶液,分别精密量上述各溶液10μl,注入液相色谱仪,记录色谱图,具体方法见表5。在表5中提及的溶剂为【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】。
[0072]
表5破坏试验结果
[0073][0074][0075]
结果表明,本品在酸、碱、高温、氧化、光照条件下,各降解产物与主峰的分离度良
好,各破坏样品主峰纯度符合要求。物料平衡以未破坏样品总峰面积/称样量为100%,其它破坏样品总峰面积/各自称样量与未破坏比值进行物料平衡计算,均在95%-105%之间,物料守恒。故此条件适合于本品有关物质检测。
[0076]
2.3定量限、检测限的确定
[0077]
分别取右旋雷贝拉唑钠对照品及杂质2、3、4、5、6、7、8、9、10、20、21、22各适量,配制一定浓度的样品,逐步稀释后精密量取10μl,注入液相色谱仪,记录色谱图,以信噪比s/n=3、s/n=10测定,结果见表6。
[0078]
表6检测限及定量限结果
[0079][0080][0081]
结果表明,在本品有关物质浓度及色谱条件下,右旋雷贝拉唑钠及各已知杂质均具有适宜的检测灵敏度,含量为0.05%的各杂质均可准确定量。
[0082]
2.4样品溶液稳定性试验
[0083]
取右旋雷贝拉唑钠样品适量,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】溶解制成每1ml约含右旋雷贝拉唑钠1mg的溶液作为供试品溶液,分别在配制后室温放置0h、4h、8h、12h、16h、20h、24h进样分析,以考察样品溶液中各杂质及主成分的含量,以面积归一化法计,结果见表7。
[0084]
表7样品溶液稳定性试验(室温15℃)
[0085][0086]
结果表明:样品在室温(15℃)条件下,样品溶液24h稳定。
[0087]
2.5线性
[0088]
2.5.1杂质贮备液配制
[0089]
取右旋雷贝拉唑钠对照品及杂质2、3、4、5、6、7、8、9、10、20、21、22各适量,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】溶解并稀释制成为每1ml约含杂质2、3、5、6、7、8、9、10、20、21、22为2μg、杂质4为10μg,右旋雷贝拉唑钠对照品为20μg的溶液,作为对照品储备液,置于2-8℃条件下保存。
[0090]
2.5.2线性
[0091]
分别精密吸取对照品储备液适量,用溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释得到一系列浓度的溶液。精密吸取上述系列梯度浓度溶液各10μl,从定量限到高浓度依次进样分析,记录色谱图,以杂质对照品溶液浓度c(μg/ml)为横坐标,杂质对照品峰面积为纵坐标,进行线性回归并求出回归方程,结果见表8和图3。
[0092]
表8线性考察结果(n=5)
[0093][0094][0095]
2.6进样精密度试验
[0096]
精密称取各杂质及右旋雷贝拉唑钠对照品适量,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释制成每1ml中含各杂质10μg/ml(杂质4为5μg/ml)及右旋雷贝拉唑钠10μg/ml的混合溶液,连续进样测定6次,考察峰面积及保留时间的变化情况。结果见表9。
[0097]
表9进样精密度试验结果
[0098][0099]
结果表明,右旋雷贝拉唑钠及各杂质峰面积rsd均小于2.0%,保留时间rsd均小于1.0%,仪器进样精密度良好。
[0100]
2.7混合对照溶液稳定性
[0101]
取右旋雷贝拉唑钠对照品和各杂质对照品(杂质2、3、4、5、6、7、8、9、10、20、21、22)适量,用溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】溶解制成每1ml约各杂质10μg/ml(杂质4为5μg/ml)及右旋雷贝拉唑钠10μg/ml的混合溶液,作为对照品溶液,分别在配制后室温放置0h、4h、8h、12h、16h、20h、24h进样分析,结果见表10。
[0102]
表10杂质对照溶液稳定性试验结果(室温15℃)
[0103][0104]
结果表明,杂质对照溶液配制后室温放置24小时内稳定。
[0105]
2.8重复性试验
[0106]
取右旋雷贝拉唑钠样品适量,精密称取6份,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】溶解并分别制成每1ml中含1mg的溶液作为供试品溶液。精密量取上述溶液各10μl,分别注入液相色谱仪,按外标法以峰面积计算供试品中各已知杂质的含量,以主成分对照法计算供试品溶液中未知杂质的含量,测定结果见表11。
[0107]
表11重复性试验结果
[0108][0109][0110]
注:
“‑”
表示未检出,含量小于0.02%不计算rsd值。
[0111]
2.9回收率试验
[0112]
精密称取右旋雷贝拉唑钠样品九份,每份约10mg,至10ml容量瓶中,分别加入杂质限量的80%、100%、120%的杂质对照,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释至刻度,分别精密量取10μl,注入液相色谱仪,进行回收率测定,结果见表12~表23。
[0113]
表12杂质2回收率试验结果
[0114][0115]
表13杂质3回收率试验结果
[0116][0117]
表14杂质4回收率试验结果
[0118][0119][0120]
表15杂质5回收率试验结果
[0121][0122]
表16杂质6回收率试验结果
[0123][0124]
表17杂质7回收率试验结果
[0125][0126]
表18杂质8回收率试验结果
[0127][0128][0129]
表19杂质9回收率试验结果
[0130][0131]
表20杂质10回收率试验结果
[0132][0133]
表21杂质20回收率试验结果
[0134][0135][0136]
表22杂质21回收率试验结果
[0137][0138]
表23杂质22回收率试验结果
[0139][0140]
2.10中间精密度试验
[0141]
取右旋雷贝拉唑钠样品6份,分别加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:
60)】溶解并稀释制成每1ml约含右旋雷贝拉唑钠为1mg的溶液,作为供试品溶液。精密量取10μl,分别注入液相色谱仪,记录色谱图。按按外标法以峰面积计算供试品溶液中各已知杂质的含量,按主成分对照法计算供试品溶液中未知杂质的含量,结果见表24。
[0142]
表24中间精密度试验结果
[0143][0144][0145]
注:仪器1:高效液相色谱仪1100,编号:100141a;仪器2:高效液相色谱仪1100,编号:100129a。
[0146]
试验结果表明:本方法中间精密度良好。
[0147]
2.11校正因子测定
[0148]
在2台液相色谱仪上,使用3根色谱柱,以p
32
排列组合,共计6次,在定量限至规定限量浓度的200%内,共制备6份样品,以各成分浓度对各峰面积进行回归,并计算杂质相对右旋雷贝拉唑钠的校正因子,结果见表25。
[0149]
表25校正因子的测定
[0150]
[0151]
[0152]
[0153][0154]
注:色谱柱1:wondasil c18-wr柱(250mm
×
4.6mm,5μm),s/n:7k5707-21;
[0155]
色谱柱2:wondasil c18-wr柱(250mm
×
4.6mm,5μm),s/n:5f5701-11;
[0156]
色谱柱3:wondasil c18-wr柱(250mm
×
4.6mm,5μm),s/n:6j5702-14;
[0157]
仪器a:agilent1100高效液相色谱仪,仪器编号100137a;
[0158]
仪器b:agilent1100高效液相色谱仪,仪器编号100141a。
[0159]
试验结果表明:杂质2的校正因子为0.57,杂质3的校正因子为0.88,杂质4的校正因子为1.13,杂质5的校正因子为0.71,杂质6的校正因子0.67,杂质7的校正因子为0.74,杂质8的校正因子为0.88,杂质9的校正因子为0.93,杂质10的校正因子为0.69,杂质20的校正因子为0.76,杂质21的校正因子为0.81,杂质22的校正因子为0.81。
[0160]
2.12耐用性考察
[0161]
为考察本方法对条件发生微小变化时的耐受程度,进行耐用性试验。考察因素包括:有机相起始比例变化25
±
2%,流动相ph7.0
±
0.2,柱温20
±
2℃及不同色谱柱试验。考察指标包括供试品中主成分保留时间、主峰与相邻峰的分离度(最小分离度)、理论板数、检
出杂质峰个数、杂质含量(已知杂质按主成分对照加校正因子法计算),测定结果见表26~表29。
[0162]
2.12.1流动相初始有机相比例的变化
[0163]
分别考察了初始有机相(甲醇)比例为23%、25%、27%条件下耐用性,测定结果见表26。
[0164]
表26耐用性考察(流动相初始有机相比例)
[0165][0166]
结果表明:初始有机相(甲醇)比例在23%~27%范围内变化时,主峰保留时间、理论板数和分离度发生明显变化,其它色谱行为及样品检测结果无明显变化。
[0167]
2.12.2流动相ph值的变化
[0168]
分别考察了流动相ph值为6.8、7.0、7.2条件下耐用性,测定结果见表27。
[0169]
表27耐用性考察(流动相ph值)
[0170][0171]
结果表明:流动相的ph值在6.8~7.2范围内变化时,主峰保留时间、理论板数和分离度有明显变化,其他色谱行为及样品检测结果均无明显变化。
[0172]
2.12.3柱温的变化
[0173]
为考察柱温对色谱行为的影响,分别考察柱温为18℃、20℃、22℃的耐用性,测定结果见表28。
[0174]
表28耐用性考察(柱温)
[0175][0176]
结果表明:柱温在18℃~22℃范围内变化时,主峰保留时间和理论板数和分离度
有明显变化,其它色谱行为及样品检测结果无明显变化。
[0177]
2.12.4色谱柱的变化
[0178]
分别考察了不同色谱柱的耐用性,测定结果见表29。
[0179]
表29耐用性考察(不同色谱柱)
[0180][0181]
注:色谱柱1为wondasil c18-wr柱(250mm
×
4.6mm,5μm),s/n:6j5702-14;
[0182]
色谱柱2为wondasil c18-wr柱(250mm
×
4.6mm,5μm),s/n:5f5701-11。
[0183]
色谱柱3为wondasil c18-wr柱(250mm
×
4.6mm,5μm),s/n:7k5707-21;
[0184]
色谱柱1和3的杂质个数12,是由于杂质2含量小于0.005%,未列入计算范围。
[0185]
结果表明:不同批次的色谱柱,对色谱行为及样品检测结果无影响。
[0186]
综上所述,本发明采用高效液相色谱法,分别从色谱条件等方面进行筛选、优化,拟定有关物质方法,进行方法学验证,对杂质1(sma)进行定性研究、对杂质2(smb)、杂质3(中间体1)、杂质4~10,20~22(工艺杂质及降解杂质)进行了定量研究。本发明的检测方法,右旋雷贝拉唑钠及有关物质线性关系良好,准确度和精密度均良好,专属性强,稳定性高。本检测方法的重现性好,能满足右旋雷贝拉唑钠原料药有关物质的检测要求,可用于右旋雷贝拉唑钠原料药的质量控制。
[0187]
对比例1不同梯度在色谱分析过程中对检测结果的影响
[0188]
液相色谱条件:色谱柱采用岛津wondasil c
18-wr柱(250x4.6mm,5μm);以20mmol/l磷酸二氢钾缓冲溶液(采用氢氧化钠调节其ph至7.0)为流动相a,以甲醇为流动相b,按下表30进行梯度洗脱;流速为1.0ml/min;柱温20℃;检测波长为280nm。
[0189]
表30梯度洗脱过程
[0190][0191]
分别取杂质1、2、3、4、5、6、7、8、9、10、20、21、22和右旋雷贝拉唑钠样品适量,置10ml量瓶中,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释至刻度,使得杂质2、3、5、6、7、8、9、10、20、21和22为1μg/ml(其中,杂质1为10μg/ml,杂质4为5μg/ml,主成分的浓度为1mg/ml),摇匀,作为杂质与样品混合溶液。
[0192]
取上述溶液10μl注入液相色谱仪,记录色谱图。
[0193]
表31不同梯度洗脱条件对色谱分析结果的影响
[0194][0195]
由图4和表31可知,该梯度洗脱过程中,在杂质与样品混合溶液中,各杂质间与主成分均能较好的分离,但44.425min处杂质1与未知杂质1(44.718min处)存在互相干扰,分离度为0.78,45.993min处的未知杂质2与46.314min处未知杂质3分离度为0.83,均未达到基线分离。
[0196]
对比例2不同柱温在色谱分析过程中对检测结果的影响
[0197]
液相色谱条件:色谱柱采用岛津wondasil c
18-wr柱(250x4.6mm,5μm);以20mmol/l磷酸二氢钾缓冲溶液(采用氢氧化钠调节其ph至7.0)为流动相a,以甲醇为流动相b,按下表30进行梯度洗脱;流速为1.0ml/min;柱温25℃;检测波长为280nm。具体洗脱过程见表1。
[0198]
分别取杂质1、2、3、4、5、6、7、8、9、10、20、21、22和右旋雷贝拉唑钠样品适量,置10ml量瓶中,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释至刻度,使得杂质2、3、5、6、7、8、9、10、20、21和22为1μg/ml(其中,杂质1为10μg/ml,杂质4为5μg/ml,主成分的浓度为1mg/ml),摇匀,作为杂质与样品混合溶液。
[0199]
取上述溶液10μl注入液相色谱仪,记录色谱图。
[0200]
表32不同柱温对色谱分析结果的影响
[0201][0202]
由图5和表32可知,该柱温的条件下,在杂质与样品混合溶液中,未知杂质3(53.038min)与杂质22(53.815min)间的分离度为1.36,未达到基线分离。
[0203]
对比例3不同色谱柱在色谱分析过程中对检测结果的影响
[0204]
液相色谱条件:色谱柱采用岛津inertsil ods-3柱(250x4.6mm,5μm);以20mmol/l磷酸二氢钾缓冲溶液(采用氢氧化钠调节其ph至7.0)为流动相a,以甲醇为流动相b,按下表30进行梯度洗脱;流速为1.0ml/min;柱温20℃;检测波长为280nm。具体洗脱过程见表1。
[0205]
分别取杂质1、2、3、4、5、6、7、8、9、10、20、21、22和右旋雷贝拉唑钠样品适量,置10ml量瓶中,加溶剂【0.01mol/l氢氧化钠-甲醇(体积比为40:60)】稀释至刻度,使得杂质2、3、5、6、7、8、9、10、20、21和22为1μg/ml(其中,杂质1为10μg/ml,杂质4为5μg/ml,主成分的浓度为1mg/ml),摇匀,作为杂质与样品混合溶液。
[0206]
取上述溶液10μl注入液相色谱仪,记录色谱图。
[0207]
表33不同柱温对色谱分析结果的影响
[0208][0209]
由图6和表33可知,该色谱柱的条件下,在杂质与样品混合溶液中,杂质20(33.563min)与主峰(34.076min)的分离度为1.19,未达到基线分离。
[0210]
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可能对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
