1.本发明属于化合物的数据处理技术领域,尤其是涉及一种色谱质谱分析用数据处理方法,其对利用由液相色谱仪与质谱分析装置组合而成的液相色谱质谱分析装置(lc-ms)、由气相色谱仪与质谱分析装置组合而成的气相色谱质谱分析装置(gc-ms)、或者毛细管电泳仪与质谱分析装置组合而成的毛细管电泳质谱分析装置(ce-ms)等色谱质谱分析装置收集到的数据进行处理,用以对试样中所含的化合物进行鉴定或结构推断。
背景技术:
2.色谱技术作为复杂组分分离和定量分析的手段之一,具有分析速度快、分离效率高、样品用量少等优势。应用色谱技术分离复杂成分,在理想实验条件下,每个单峰对应一个组分,但实际上,当两个或多种成分含有相似的结构或性质时,很容易形成重叠峰,在准确定量研究中,采用传统的方法如切线法和垂直切割法等,虽然快速但精度低,这为后续的定性和定量分析带来困难。因此,如何有效分离重叠色谱峰,是目前亟需解决的重要问题之一。
3.进几十年来不断完善的化学计量学方法能较为准确的解析重叠的色谱峰,比如纵向迭代法,该方法从远离重叠区域的前后沿开始拟合,对色谱峰重叠区域进行修正,但该方法要求重叠峰具有谷点;基于代数的分峰方法主要有基于高斯函数的谱峰拟合算法、基于最小二乘法的谱峰拟合算法、小波变换算法等。这些法能够得到较好的计算结果和分峰效果,然而在分峰过程中,它们需要一定的参数估计与优化、模型选择等步骤,计算量大,消耗时间长,不适用于实时在线处理。基于模式识别的分峰方法中,免疫算法分离效果较好,但它仅适用于已知成分的重叠峰分离,应用受限。
4.质谱是一种高效、灵敏的技术,能提供化合物的结构信息。但是,对于复杂的化合物,如糖类,由于存在微观不均一性,化合物种类繁多、分离困难、质谱信号多且重叠、带有多种电荷等,因此,对于复杂的化合物,大量质谱数据的高效准确处理将形成高通量组学研究的瓶颈。一方面,手工标注这些分析数据既耗时又效率低下,更大的问题是缺乏准确性和标准,丰度低、数据误差大的峰容易漏检。一方面,目前出现了一系列辅助糖结构信息分析的软件和方法,速度快、操作简单,数据库的建立大大降低了繁琐的数据处理带来的难度,通过数据库的搜索和分析得到的数据全面、准确且不遗漏,将根据同位素峰的系列进行分级,更直观地反映峰归属的可信度。这些软件和方法为新糖分子的发现、高通量高效研究以及糖质谱数据的注释鉴定提供有利支持。
5.然而由于寡糖结构的复杂性,现有的研究工作对寡糖质谱形成机理的认识还存在局限性。因此,理论质谱预测的准确度不高,影响分析结果的准确性,在得到的数据中存在大量的假阳性结果。一类假阳性数据是由于质谱信号重叠严重,电荷识别错误,从而去卷积错误,最终导致归属的组成错误,这类假阳性数据的分子量与保留时间不相符。一类假阳性数据是由于硫酸寡糖在离子源中容易丢失磺酸基,所以对于低硫酸化程度的寡糖来说,质谱在归属的时候很难将其与高硫酸化寡糖丢失磺酸基产生的碎片离子峰区分开,在解析时
没有办法给出确定糖链组成。
6.综上所述,对于复杂样品体系的色谱中存在的大量重叠峰,其定性和定量分析均非常困难。对于质谱数据库检索结果,分析人员人工解析、确认、判断、鉴定化合物,对于分析人员而言是较大的负担。在对医药品或非法药物开展分析时,尤其是在分析大量基本结构骨架相同、取代基等存在些许差异的复杂化合物体系过程中,有效的、准确的、自动化的液质联用分析、检索、解析手段和工具就显得尤为重要。
技术实现要素:
7.本发明目的是为了解决上述技术问题,提供一种色谱质谱分析用数据处理方法,通过对色谱、质谱分析数据进行自动化处理来准确判断复杂样品中多种化合物的组成比例和每种组分的结构组成,因此不再需要分析人员自己对数据库检索结果进行确认或比较的麻烦操作,从事鉴定作业的分析人员的负担可大幅减轻。
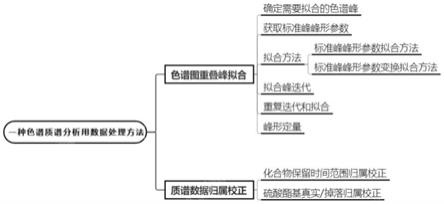
8.本发明的技术方案是:一种色谱质谱分析用数据处理方法,通过对色谱、质谱数据进行分析处理来确定复杂样品中多种化合物的组成,包括对色谱图进行重叠峰拟合,具体步骤如下:
9.步骤1:确定需要拟合的色谱峰;
10.步骤2:采用相同色谱条件分离可基线分离的化合物得到标准色谱峰,通过每个标准色谱峰的左右标准差获得标准色谱峰峰形参数;
11.步骤3:根据标准色谱峰峰形参数对确定需要拟合的色谱峰进行第一次拟合;
12.步骤4:对单个区域色谱峰进行迭代并拟合,然后对完整区域色谱峰进行迭代并拟合;
13.步骤5:重复上述完整区域色谱峰迭代和拟合过程,并将完整色谱峰的拟合峰进行叠加,将叠加峰与原始色谱峰数据进行拟合度r2计算,当拟合度r2达到最大时,停止迭代计算;
14.步骤6:将最后一次迭代计算出的单个拟合峰峰形曲线用于该区域保留时间确认、积分和峰形分析,完成从多重峰色谱图中剥离出单独峰。
15.作为优选的技术方案,步骤4中对单个区域色谱峰进行迭代并拟合,具体方法如下:
16.步骤41:在某个区域内的n个色谱峰,用原始色谱峰数据减去其它区域的色谱峰以及该区域内p1…
p
n-1
的叠加峰后,调整pn的顶点位置并拟合;
17.步骤42:用原始色谱峰数据减去其它区域内色谱峰以及该区域内p1…
p
n-2
、pn的叠加峰后,调整p
n-1
的顶点位置并拟合,直到对该区域内的n个色谱峰的位置均进行调整;
18.步骤43:将n个色谱峰的拟合峰进行叠加,将叠加峰与原始色谱峰数据进行对比,根据对比差距进行峰形参数的调整并拟合,当该区域局部拟合度r2达到最大,停止迭代。
19.作为优选的技术方案,步骤4中对完整区域色谱峰进行迭代并拟合,具体方法如下:
20.按照从左往右、或从右往左的顺序,从一个区域迭代拟合到下一个区域迭代拟合,直到所有区域色谱峰完成迭代,即完成一次完整区域色谱峰迭代和拟合。
21.作为优选的技术方案,还包括根据化合物保留时间范围进行质谱归属校正,其具
体方法如下:
22.步骤1:建立复杂体系所有可能化合物准确分子量数据库;
23.步骤2:将质谱数据中质荷比去卷积,得到相应的准确分子量;与建立的数据库中对应结构特征的理论分子量进行匹配,若实际分子量与理论分子量的偏差小于20ppm,完成第一次归属;
24.步骤3:根据色谱拟合结果中每组化合物的保留时间分布范围进行第二次归属,如果化合物的结构特征与保留时间相符,则确认归属;如果化合物的结构特征与保留时间不相符,则归属错误,将归属错误质谱信号再按照不同电荷数重新去卷积,所得准确分子量的结构特征与色谱拟合中相应保留时间进行匹配,直至所有可能匹配结束,确认所有归属并剔除无法归属质谱信号,即假阳性信号。
25.作为优选的技术方案,还包括对硫酸酯基真实/掉落进行归属校正,其具体方法如下:
26.根据硫酸酯基在质谱离子源内掉落形成的低硫酸化化合物以及真实的低硫酸化化合物与高硫酸化化合物的保留时间差异,对硫酸酯基真实/掉落进行归属校正。
27.作为优选的技术方案,所述色谱分析数据为采用色谱、电泳或者其它分离技术,对复杂化合物进行分离分析的色谱图或者电泳转化的色谱图。
28.作为优选的技术方案,所述质谱分析数据为当色谱、毛细管电泳或者其它分离技术与质谱联用时,所得到的质荷比、峰度、强度、同位素信号、总离子流(tic)或者上述数据经过数据库检索得到的化合物组成数据。
29.作为优选的技术方案,步骤1中通过色谱峰的一阶导数和二阶导数确定需要拟合的色谱峰,且该色谱峰包括正常峰、肩峰以及隐藏峰。
30.本发明的优点是:
31.1.本发明的色谱质谱分析用数据处理方法,通过对色谱、质谱分析数据进行自动化处理来准确判断复杂样品中多种化合物的组成比例和每种组分的结构组成,因此不再需要分析人员自己对数据库检索结果进行确认或比较的麻烦操作,从事鉴定作业的分析人员的负担可大幅减轻;
32.2.本发明可以根据色谱图拟合峰进行相对定量和质谱定性分析,可确认每个测试样品中真实的化合物组成,从而进一步探索机制、构效关系等。
附图说明
33.为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
34.下面结合附图及实施例对本发明作进一步描述:
35.图1为本发明色谱质谱分析用数据处理方法的流程图;
36.图2为本发明三类拟合峰的特征示意图;
37.图3为本发明化合物保留时间范围归属校正示意图;
38.图4为本发明硫酸酯基真实/掉落归属校正示意图;
39.图5为本发明在实施例1中针对化合物种类未知且无法得知标准峰形的色谱图重
叠峰拟合和化合物保留时间范围归属校正的一个应用示意图;
40.图6为本发明在实施例2中针对明确化合物种类且有标准峰形的色谱图重叠峰拟合的一个应用示意图;
41.图7为本发明在实施例3中针对化合物保留时间范围归属校正和硫酸酯基真实/掉落归属校正的一个应用示意图。
具体实施方式
42.以下结合具体实施例对上述方案做进一步说明。应理解,这些实施例是用于说明本发明而不限于限制本发明的范围。实施例中采用的实施条件可以根据具体厂家的条件做进一步调整,未注明的实施条件通常为常规实验中的条件。
43.实施例1
44.实验目的:采用高分辨液质联用,分析依诺肝素钠寡糖序列,从而进行不同动物来源的依诺肝素钠样品的一致性评价。
45.实验方法:利用串联两种不同孔径的分子筛色谱柱进行分离,同时结合高分辨质谱对依诺肝素钠寡糖进行分析。因依诺肝素钠寡糖非还原端有双键,在232nm处有特征吸收,因此本实验中分析232nm处的紫外谱图。通过glycresoft建立依诺肝素钠的数据库,通过数据库检索获得质谱数据,检索结果中存在大量假阳性结果。
46.数据处理方法(参照图1):
47.1、色谱图重叠峰拟合:
48.1)确定需要拟合的色谱峰:通过色谱峰的一阶导数和二阶导数来确定需要拟合的色谱峰。根据正常峰、肩峰及隐藏峰三类峰的特征确定需要拟合的色谱峰,参照图2所示,其中正常峰:有明显的峰顶点,峰顶点的一阶导数为0,且峰顶点左边一阶导数大于0,右边小于0;对于肩峰,则会引起一阶导数的改变,出现极值点;对于隐藏峰,则相当于在一阶导数上形成肩峰,则需要根据二阶导数来确定隐藏峰的位置。
49.2)获取标准峰峰形参数:采用相同的色谱条件分离可基线分离的化合物,得到标准色谱峰1(窄峰)和标准色谱峰2(宽峰)。通过公式1:对标准色谱峰进行拟合,分别得到了每个标准色谱峰的左右标准差(峰形参数),如标准色谱峰1的左右标准差分别为σ
a1
和σ
a2
,标准色谱峰2的左右标准差分别为σ
b1
和σ
b2
,色谱峰由有数个等间距点组成,公式中x,y分别为每个点对应的时间和响应,h为该色谱峰的峰高,t为该色谱峰的保留时间,σ为该色谱峰的标准差(一般为峰高0.607倍处的峰宽之半)。
50.3)第一次拟合:根据标准色谱峰2的左右标准差带入上述公式1对确定需要拟合的色谱峰进行第一次拟合。
51.4)拟合峰迭代:
52.a)单个区域迭代和拟合:在某个区域的n个色谱峰中,用原始色谱峰数据减去其它区域色谱峰以及该区域p1…
p
n-1
的叠加峰后,调整pn的顶点位置并拟合;用原始色谱峰数据减去其它区域色谱峰以及该区域p1…
p
n-2
、pn的叠加峰后,调整p
n-1
的顶点位置并拟合
……
以此类推,对该区域的n个色谱峰的位置均进行调整。然后将n个色谱峰的拟合峰进行叠加,将叠加峰与原始色谱峰数据进行对比(局部拟合度r2),根据对比差距进行峰形参数调整并拟
合,当该区域局部拟合度r2达到最大,停止迭代。
53.b)完整色谱峰迭代和拟合:从最右边区域(即dp2)开始进行迭代和拟合,从右往左进行下一个区域的迭代,直到所有区域完成迭代,为一次完整色谱峰迭代和拟合。
54.c)重复迭代和拟合:不断重复以上完整色谱峰迭代和拟合过程,将拟合峰进行叠加,将叠加峰与原始色谱峰数据进行拟合度r2计算,当拟合度r2达到最大时,停止迭代计算。
55.5)峰形定量:将最后一次迭代计算出的单个拟合峰峰形曲线用于定量和峰形分析,从而完成从多重峰色谱图中剥离出单独峰的过程。
56.2、质谱数据归属校正(参照图3所示,针对化合物保留时间范围归属校正):
57.根据拟合的结果,得到了每个化合物组的保留时间分布范围。接下来利用化合物的结构特征和保留时间分布范围对检索结果中匹配的寡糖组成进行验证。从而将数据分为两类,一类为归属正确的;一类为归属错误的,重新去卷积,即按照组成的保留时间确定其正确归属的结构特征,再根据质荷比重新去卷积,得到不同电荷(1-10电荷)对应的分子量,再与建立的依诺肝素钠数据库中对应结构特征的理论分子量进行匹配,如计算分子量与理论分子量偏差小于20ppm,则校正归属。
58.一致性评价(参照图5所示):将不同动物来源的依诺肝素钠经过上述数据处理方法后,将每个测试样品最终的寡糖组成进行分析,评价依诺肝素钠样品的一致性。从pca分析图中可看出,经过假阳性寡糖数据的去除,可成功将猪肠来源的依诺肝素钠与其他动物来源的依诺肝素钠样品区分开。
59.实施例2
60.实验目的:肝素类药物中可能存在其他杂质多糖,采用高效液相色谱分离不同糖胺聚糖,测得肝素类药物生成过程中其他杂质多糖的含量。
61.实验方法:利用阴离子交换色谱柱分离,测试不同浓度的混合标准品(图中2为肝素,为主要成分),经过拟合得到各自的峰面积,得到不同糖胺聚糖的线性关系。
62.数据处理方法(参照图1和图6):
63.1.确定需要拟合的色谱峰。
64.2.获取标准峰峰形参数:将重叠峰中各个化合物单独测试,获得每个化合物的色谱图,通过公式对标准色谱峰进行拟合,分别得到了每个标准色谱峰的左右标准差。
65.3.第一次拟合:将每个标准色谱峰的峰形参数代入公式1,相应地对确定的需要拟合的色谱峰进行拟合。
66.4.拟合峰迭代:用多重峰的峰形减去第一次拟合得到的单独峰2、3、4的叠加峰形,第一次迭代出单独峰1的峰形;用多重峰的峰形减去第一次拟合得到的单独峰1、3、4的叠加峰形,第一次迭代出单独峰2的峰形;......;
67.5.重复迭代和拟合:不断重复以上3)和4)的迭代和拟合过程,将各个拟合峰进行叠加,将叠加峰与原始多重峰进行r2计算,当r2达到最大时停止迭代计算。
68.6.峰形定量:使用最后一次迭代计算出的单个拟合峰峰形曲线用于定量。测试了7个浓度下的混合标准品(0.5、2、4、10、15、20、50mg/ml),并且拟合得到了各个化合物的不同浓度下的峰面积,得到了各个化合物的线性关系。
69.7.实际样品定量:测试实际样品,进行拟合得到相应的峰面积,代入线性关系,得到化合物1的浓度为6.89mg/ml,化合物2的浓度为44.39mg/ml,化合物3的浓度为7.95mg/
ml,化合物4的浓度为7.43mg/ml,因此,各成分的含量分别为10.3%、66.6%、11.9%和11.1%。
70.实施例3
71.实验目的:采用高分辨液质联用,分析卡拉胶酸解不同时间的酸解产物的寡糖序列,进行卡拉胶酸解规律的探究。
72.实验方法:利用串联两种不同孔径的分子筛色谱柱分离,同时结合高分辨质谱对卡拉胶酸解不同时间的酸解产物进行寡糖分析。因卡拉胶无紫外特征吸收,本实验不进行紫外色谱图分析。采用glycresoft建立卡拉胶寡糖的数据库,通过数据库检索获得质谱数据,同样检索结果中存在大量假阳性结果。
73.数据处理方法:
74.1、质谱数据归属校正(参照图3所示,针对化合物保留时间范围归属校正):手动解析质谱图确定了每个化合物组的时间分布范围。接下来利用化合物的结构特征和保留时间的关系对匹配到的寡糖组成归属进行验证。从而将数据分为两类,一类为归属正确的;一类为归属错误的,重新去卷积,即按照组成的保留时间确定其正确归属的结构特征,再根据质荷比重新去卷积,得到不同电荷对应的分子量,再与建立的卡拉胶数据库中对应结构特征的理论分子量进行匹配,如计算分子量与理论分子量偏差小于20ppm,则校正归属。
75.2、硫酸酯基真实/掉落归属校正(参照图4):首先根据分离方法的特性(体积排阻色谱,分子量大的先出峰),在单个结构特征所属范围内,高硫酸化寡糖丢失磺酸基产生的碎片离子峰与真实的低硫酸化寡糖分子量相同,但保留时间有差异;硫酸酯基掉落形成的低硫酸化寡糖与高硫酸化寡糖保留时间接近(<0.2min),而真实的低硫酸化寡糖与高硫酸化寡糖保留时间间隔大。而且,各个结构特征所属范围内,糖基组成相同仅硫酸化程度不同的寡糖组成呈线性关系。
76.酸解规律探究,参照图7,其中eic图中虚线代表偶数寡糖,实线代表奇数寡糖:
77.在相同的终止ph条件(ph=1)下,卡拉胶酸解不同时间(1h、3h、6h、12h),可发现酸解不同时间的酸解产物主要均为偶数糖。酸解1h的酸解产物的聚合度为dp2~dp38,且主要为硫酸化程度最高的寡糖组成;酸解3h、6h和12h的酸解产物均存在多个真实的低硫酸化的寡糖组成。
78.在相同的酸解时间(3h),不同的终止ph条件(ph=1、7、9和12)下,可发现ph=1、7的酸解产物主要为偶数糖;ph=9的酸解产物偶数糖和奇数糖均存在;ph=12的酸解产物主要为奇数糖。
79.因此,可发现相同的ph终止条件,随着酸解时间延长,卡拉胶寡糖聚合度逐渐降低;而且相同的酸解时间,随着酸性条件向碱性条件过渡,卡拉胶奇数寡糖的含量逐渐增加。
80.上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。