1.本发明涉及临床质谱体外诊断试剂领域,特别涉及一种基于磁性固相萃取的儿茶酚胺及其代谢物的液相色谱串联质谱检测方法。
背景技术:
2.儿茶酚胺类物质由肾上腺髓质、交感神经、副神经节嗜铬细胞合成和分泌,它们既是激素,又是神经递质。儿茶酚胺类物质主要包括多巴胺(da)、去甲肾上腺素(ne)和肾上腺素(e),儿茶酚胺氧甲基转移酶将da、e、ne分别转变为甲氧酪胺(3-mt)、变肾上腺素(mn)、去甲变肾上腺素(nmn)。儿茶酚胺是生物体内具有较强生理学效应的内源性物质,在脑和神经信号传导中起着重要的作用。生物体内儿茶酚胺水平与多种生理、病理现象有着密切关系。儿茶酚胺类物质对人体的心血管系统、神经系统、内分泌系统、肾脏、呼吸系统等生理活动起着广泛的调节作用。例如,肾上腺素既对心血管起作用,又是促进分解代谢的激素,多巴胺作为治疗帕金森病的特效药,能穿越血脑屏障脱羧后产生神经递质;去甲肾上腺素为拟交感神经药,具有收缩血管、升高血压的作用。此外,儿茶酚胺还与一些功能性疾病如神经分裂症、忧郁症以及器官性病变的出现相关。通过检测生物样本中的儿茶酚胺及其代谢产物水平,不仅对嗜铬细胞瘤、副神经节瘤、神经母细胞瘤、高血压、心肌梗塞、肾上腺髓质增生等疾病的诊断和治疗具有重要的临床意义,并且有助于甲状腺功能异常、充血性心力衰竭、糖尿病、肾脏功能不全等疾病的诊断。综上,生物体中儿茶酚胺及其代谢产物浓度水平的信息在神经生理研究与递质代谢异常相关疾病诊断治疗及预后有着极其重要意义。依据《嗜铬细胞瘤和副神经节瘤诊断治疗的专家共识2020版》中提出首选血浆游离或尿液甲氧基肾上腺素类浓度测定,建议可同时检测血或尿ne、da、e及其代谢产物浓度以帮助诊断嗜铬细胞瘤和副神经节瘤。
3.血浆中儿茶酚胺及其代谢产物含量极低,达到pg级别,在对其定量检测方法中,最常用的是高效液相色谱和电化学检测联用法,但仪器检测灵敏度较低,无法满足临床检测需求。此外,高效液相色谱和电化学检测联用法所需的血浆样本量较大,样本前处理所需的时间长,为了在实现目标分析物的所需的检测灵敏度往往需要很长时间的色谱分离,共洗脱的干扰组分对结果的准确度存在很大的影响。基于免疫的检测方法能够检测血浆中变肾上腺素的含量,由于交叉反应和非特异性的结合会导致检测准确度低,且不同的免疫测试的结果可能不统一。由于许多分析方法在灵敏度、选择性及分析速度等方面达不到血浆中儿茶酚胺及其代谢产物定量分析的要求,限制了其在临床检测上进一步的推广和开展。
4.以色谱作为分离手段,三重四极杆质谱作为检测手段的色谱-质谱联用技术成为了定量检测生物样品中痕量目标分析物的强有力的手段,因此被用于血浆中儿茶酚胺及其代谢产物的定量检测。专利cn109142594a报道了一种利用液相色谱串联质谱检测体液样本中儿茶酚胺和变肾上腺素的方法,该方法需要先对样品进行蛋白沉淀,移取上清液使用n2吹干后加入pitc进行衍生化反应,衍生化产物使用n2吹干后加入复溶剂得到待测样本,使
用液相色谱串联质谱进行检测衍生化,前处理繁琐,耗费时间长,检测效率低。专利cn111896643a公开一种人血浆中儿茶酚胺的液相色谱质谱联用的检测方法,血浆样本采用离子交换固相萃取进行前处理,色谱柱采用反相五氟苯基(f5)色谱柱,为反相作用机理色谱柱。该方法样品前处理需要大量人工参与,极大降低实验效率,同时由于儿茶酚胺及其代谢物为强极性化合物,在f5柱上保留能力很弱,实验中色谱柱需要在高比例水相下对目标物才有保留,而高比例水相会降低质谱的离子化效率,同时会有大量弱保留的内源性干扰组分共流出,对目标物的定量造成基质干扰。目前儿茶酚胺及其代谢物检测常用的离子交换固相萃取法使用的固相萃取96孔板为高成本实验耗材,实验中固相萃取填料接触到有机试剂之后会产生溶胀现象,会影响溶液通过固相萃取填料的流速,从而产生孔间均一性差的问题。
5.所以,现在有必要提供一种更可靠的方案。
技术实现要素:
6.本发明所要解决的技术问题在于针对上述现有技术中的不足,提供一种基于磁性固相萃取的儿茶酚胺及其代谢物的液相色谱串联质谱检测方法。本发明使用了磁性固相萃取技术进行血浆中儿茶酚胺及其代谢物的前处理,其机理为基质分散型固相萃取,本发明中可使用外部磁场(如磁性固相萃取仪器、磁棒)将具有磁性的固相萃取填料进行转移,整个流程易于实现自动化,同时还保留了固相萃取高选择性、高净化率等优点。本发明中使用的磁性固相萃取填料为mg级别,耗材价格低,可降低实验耗材成本;本发明还公开一种使用基于氨基官能团的色谱柱进行目标物的检测的策略,该色谱柱为亲水作用机理,在高比例有机相下色谱柱对儿茶酚胺及其代谢物的保留能力反而更强,对目标物的离子化效率有极大提高,同时色谱柱对e(肾上腺素)、nmn(去甲变肾上腺素)这两种有共同碎片的化合物达到了基线分离,这对目标物的准确定量至关重要。
7.为实现上述目的,本发明采用的技术方案是:一种基于磁性固相萃取的儿茶酚胺及其代谢物的液相色谱串联质谱检测方法,该方法包括以下步骤:
8.1)制备检测样:
9.1-1)取待测样本,向其中加入内标溶液和水溶液,混匀;
10.1-2)将磁性固相萃取填料加入到乙腈中进行填料活化,然后将磁性固相萃取填料转移至水中进行填料平衡;
11.1-3)将步骤1-2)得到的产物中的磁性固相萃取填料转移步骤1-1)中得到的混合液中,混合;
12.1-4)将步骤1-3)得到的产物中的磁性固相萃取填料转移至水溶液中,混合;
13.1-5)将步骤1-4)得到的产物中的磁性固相萃取填料转移至乙腈中,混合;
14.1-6)将步骤1-5)得到的产物中的磁性固相萃取填料转移至乙腈与甲酸的混合液中,混合;
15.1-7)去除步骤1-6)得到的产物中的磁性固相萃取填料,剩余的溶液作为检测样;
16.2)构建目标物的标准曲线:
17.配置一定浓度梯度的儿茶酚胺及其代谢物的标准液,通过液相色谱串联质谱法检测,构建得到儿茶酚胺及其代谢物的标准曲线;
18.3)采用液相色谱串联质谱法对检测样进行检测,结合构建的标准曲线计算得到检测样中的儿茶酚胺及其代谢物的含量;
19.其中,儿茶酚胺包括多巴胺、去甲肾上腺素和肾上腺素,儿茶酚胺的代谢物包括甲氧酪胺、变肾上腺素和去甲变肾上腺素。
20.优选的是,所述步骤1-6)中,乙腈与甲酸的混合液中还含有5-20μg/ml的维生素c。
21.优选的是,所述步骤1-6)中,乙腈与甲酸的混合液中还含有10μg/ml的维生素c。
22.优选的是,乙腈与甲酸的混合液中,甲酸的体积百分数为0.5-5%。
23.优选的是,乙腈与甲酸的混合液中,甲酸的体积百分数浓度为1%。
24.优选的是,其中,采用磁性固相萃取仪器转移磁性固相萃取填料。
25.优选的是,所述步骤1)具体包括:
26.1-1)取250-450μl待测样本,向其中加入5-20μl内标溶液和200-600μl水溶液,500-2000转/min混匀1-4min;
27.1-2)将1-6mg磁性固相萃取填料加入到100-400μl乙腈中进行填料活化,然后将磁性固相萃取填料转移至水中进行填料平衡;
28.1-3)将步骤1-2)得到的产物中的磁性固相萃取填料转移步骤1-1)中得到的混合液中,混合60-240s;
29.1-4)将步骤1-3)得到的产物中的磁性固相萃取填料转移至200-600μl水溶液中,混合30-120s;
30.1-5)将步骤1-4)得到的产物中的磁性固相萃取填料转移至200-600μl乙腈中,混合30-120s;
31.1-6)将步骤1-5)得到的产物中的磁性固相萃取填料转移至50-200μl的乙腈、甲酸、维生素c的混合液中,混合30-120s;该混合液中甲酸的体积百分数浓度为1%;
32.1-7)将步骤1-6)得到的产物中的磁性固相萃取填料转移至步骤1)的产物中剩余的溶液中,丢弃;将步骤1-6)得到的产物中剩余的溶液作为检测样。
33.优选的是,所述步骤1)具体包括:
34.1-1)取390μl待测样本,向其中加入10μl内标溶液和400μl水溶液,1000转/min混匀2min;
35.1-2)将3mg磁性固相萃取填料加入到200μl乙腈中进行填料活化,然后将磁性固相萃取填料转移至水中进行填料平衡;
36.1-3)将步骤1-2)得到的产物中的磁性固相萃取填料转移步骤1-1)中得到的混合液中,混合120s;
37.1-4)将步骤1-3)得到的产物中的磁性固相萃取填料转移至400μl水溶液中,混合60s;
38.1-5)将步骤1-4)得到的产物中的磁性固相萃取填料转移至400μl乙腈中,混合60s;1-6)将步骤1-5)得到的产物中的磁性固相萃取填料转移至100μl的乙腈、甲酸、维生素c的混合液中,混合60s;该混合液中甲酸的浓度为1%;
39.1-7)将步骤1-6)得到的产物中的磁性固相萃取填料转移至步骤1)的产物中剩余的溶液中,丢弃;将步骤1-6)得到的产物中剩余的溶液作为检测样。
40.优选的是,所述步骤3)中,液相色谱检测条件为:
41.色谱柱:waters xbridge beh amide;
42.流动相:a相为含5%乙腈(v/v)的水溶液,且含浓度为10mm的乙酸铵,b相包括:含5%水(v/v)、0.1%(v/v)甲酸的乙腈溶液;
43.梯度洗脱程序:起始比例为90%b相,1.5min降至70%b相,70%b保持至3min,3.1min升至90%b相,并保持至6.6min;
44.流速:0.6ml/min;进样量:20μl;柱温:35℃。
45.优选的是,所述步骤3)中,质谱条件为:
46.离子源:电喷雾离子源;
47.检测方式:多反应监测;
48.气帘气20psi,喷雾器55psi,辅助加热气45psi,温度450℃,离子化电压3500v,碰撞气8psi。
49.本发明的有益效果是:
50.本发明提供的基于磁性固相萃取的儿茶酚胺及其代谢物的液相色谱串联质谱检测方法,通过磁性固相萃取技术对儿茶酚胺及其代谢物检测样品进行前处理,通过施加外部磁场能快速实现待测物分离、转移,便于实现自动化前处理,同时还保留了固相萃取高选择性、高净化率等优点;
51.本发明通过使用基于氨基官能团的亲水作用机理的色谱柱进行目标物的检测的策略,极大提高了质谱对目标物的离子化效率,降低目标物定量限,同时色谱柱能对e(肾上腺素)、nmn(去甲变肾上腺素)这两种有共同碎片的化合物达到基线分离,从而提高目标物的定量准确性;
52.本发明采用高比例有机相作为洗脱剂,有利于降低内源性干扰组分共流出,降低基质效应;
53.本发明通过磁性固相萃取填料分散吸附增加接触面积,能提高吸附速率及富集效果,缩短平衡时间;本发明能够避免spe流速差异、填料溶胀等误差,降低质谱检测变异性;
54.本发明的样品处理步骤能够去除内源性磷脂、蛋白质等物质的干扰,降低基质效应与仪器污染风险。
附图说明
55.图1为本发明的实施例中得到的目标物标准曲线;
56.图2为本发明的实施例中得到的目标物色谱图;
57.图3为磁性固相萃取填料转移原理示意图;
具体实施方式
58.下面结合实施例对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
59.应当理解,本文所使用的诸如“具有”、“包含”以及“包括”术语并不排除一个或多个其它元件或其组合的存在或添加。
60.试剂来源说明
61.(1)肾上腺素(e,上海谱芬生物科技有限公司);
62.(2)肾上腺素-d6(e-d6,上海谱芬生物科技有限公司);
63.(3)dl-去甲肾上腺素盐酸(ne,上海谱芬生物科技有限公司);
64.(4)dl-去甲肾上腺素-d6盐酸盐(ne-d6,上海谱芬生物科技有限公司);
65.(5)多巴胺盐酸盐(da,上海谱芬生物科技有限公司);
66.(6)多巴胺-d4盐酸盐(da-d4,上海谱芬生物科技有限公司);
67.(7)3-甲氧酪胺盐酸盐(3-mt,上海谱芬生物科技有限公司);
68.(8)3-甲氧酪胺-d4盐酸盐(3-mt-d4,上海谱芬生物科技有限公司);
69.(9)dl-去甲变肾上腺素盐酸盐(nmn,上海谱芬生物科技有限公司);
70.(10)dl-去甲变肾上腺素盐酸盐-d3(nmn-d3,上海谱芬生物科技有限公司);
71.(11)变肾上腺素盐酸(mn,上海谱芬生物科技有限公司);
72.(12)dl-变肾上腺素盐酸盐-d3(mn-d3,上海谱芬生物科技有限公司);
73.(13)乙腈(康科德科技有限公司);
74.(14)乙酸铵(thermo fisher);
75.(15)甲酸(thermo fisher);
76.(16)维生素c(sigma);
77.高纯水为符合国际gb/t 6682-2008的一级水。
78.实施例1
79.本实施例提供了一种基于磁性固相萃取的儿茶酚胺及其代谢物的液相色谱串联质谱检测方法,其中,儿茶酚胺包括多巴胺(da)、去甲肾上腺素(ne)和肾上腺素(e),儿茶酚胺的代谢物包括甲氧酪胺(3-mt)、变肾上腺素(mn)和去甲变肾上腺素(nmn)。
80.该方法包括以下步骤:
81.1)制备检测样:
82.1-1)取390μl待测样本,向其中加入10μl内标溶液和400μl水溶液,1000转/min混匀2min;
83.1-2)在干净样品管内加入3mg磁性固相萃取填料,使用200μl乙腈活化填料,用磁性固相萃取仪器将填料转移至400μl水内进行填料平衡;
84.1-3)将步骤1-2)得到的产物中的磁性固相萃取填料转移步骤1-1)中得到的混合液中,混合120s;
85.1-34)将步骤1-3)得到的产物中的磁性固相萃取填料转移至400μl水溶液中,混合60s;
86.1-5)将步骤1-4)得到的产物中的磁性固相萃取填料转移至400μl乙腈中,混合60s;
87.1-6)将步骤1-5)得到的产物中的磁性固相萃取填料转移至100μl的含甲酸体积百分数为1%的乙腈溶液(且含10μg/ml的维生素c)中,混合60s;
88.1-7)将步骤1-6)得到的产物中的磁性固相萃取填料转移至步骤1)的产物中剩余的溶液中,丢弃;将步骤1-6)得到的产物中剩余的溶液作为检测样。
89.2)构建目标物的标准曲线:
90.配置一定浓度梯度的儿茶酚胺及其代谢物的标准液,通过液相色谱串联质谱法检测,构建得到儿茶酚胺及其代谢物的标准曲线。
91.本实施例中,根据儿茶酚胺及代谢物的临床检测需求确定检测范围,并在检测范围需求内设置系列浓度梯度的儿茶酚胺及其代谢物的标准液,供液相色谱串联质谱法检测,将目标分析物和内标峰面积的比值与梯度浓度进行拟合,得到工作曲线。
92.本实施例中,使用甲醇/水(体积比1/1,且含10μg/ml维生素c)配制目标物储备液,再稀释得到标准工作液,其中e、ne、nmn标准工作液浓度为20ng/ml、50ng/ml、1000ng/ml,da标准工作液浓度为20ng/ml、100ng/ml、1000ng/ml,3-mt、mn标准工作液浓度为2ng/ml、50ng/ml、1000ng/ml。使用稀释液将目标物标准工作液进行混合配制,形成系列浓度梯度的标准液,供液相色谱串联质谱法检测,得到标准工作曲线,标准液浓度见表1,内标物工作溶液浓度为20ng/ml。
93.表1标准液浓度
[0094][0095][0096]
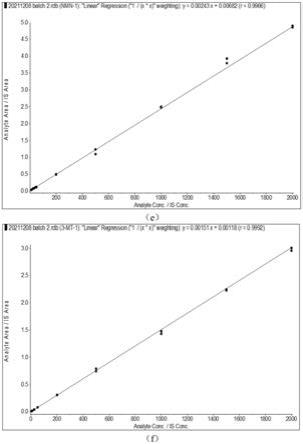
本实施例中,得到的目标物标准曲线如图1(a)-(f)所示,其中,(a)da的标准曲线;(b)e的标准曲线;(c)ne的标准曲线;(d)mn的标准曲线;(e)nmn的标准曲线;(f)3-mt的标准曲线。由图可知拟合的线性呈良好的线性关系,且r值均在0.995以上,目标物线性范围见表2。
[0097]
表2目标物线性范围及线性关系
[0098]
[0099]
3)采用液相色谱串联质谱法对检测样进行检测,结合构建的标准曲线计算得到检测样中的儿茶酚胺及其代谢物的含量;
[0100]
液相色谱检测条件为:
[0101]
1、色谱柱:waters xbridge beh amide(2.1*100mm,2.5μm);
[0102]
2、流动相:a相为含5%乙腈(v/v)的水溶液,且含浓度为10mm的乙酸铵,b相包括:含5%水(v/v)、0.1%(v/v)甲酸的乙腈溶液;
[0103]
3、梯度洗脱程序:起始比例为90%b相,1.5min降至70%b相,70%b保持至3min,3.1min升至90%b相,并保持至6.6min;
[0104]
4、流速:0.6ml/min;进样量:20μl;柱温:35℃。
[0105]
质谱条件为:
[0106]
1、离子源:电喷雾离子源(esi);
[0107]
2、检测方式:多反应监测(mrm);
[0108]
3、气帘气(cur)20psi,喷雾器(gs1)55psi,辅助加热气(gs2)45psi,温度(tem)450℃,离子化电压(is)3500v,碰撞气(cad)8psi;
[0109]
4、每种化合物的母离子、子离子、驻留时间、锥孔电压、碰撞能量等质谱参数见下表3。
[0110]
表3质谱采集参数
[0111]
q1q3dwell timeiddpepce184.216615e-136712190.2172.115e-d638613170.3152.115ne-136711176.1158.115ne-d65579198.1180.115mn-135711183.2151.215mn-d380824184.1166.115nmn-135612187.1169.115nmn-d335612168.191153-mt-144732172155.1153-mt-d443614154.191.115da-152733158.114115da-d4201030
[0112]
参照图2,为目标物色谱图,其中,(a)da的色谱图;(b)e的色谱图;(c)ne的色谱图;(d)mn的色谱图;(e)nmn的色谱图;(f)3-mt的色谱图。2b图与2e图分别为e与nmn色谱图,但是均出现了两个峰,原因为e与nmn在检测过程中有裂解现象导致两者有共同碎片,而本发明选用的色谱柱及建立的梯度条件可以将两者达到基线分离,不干扰两者的准确定量。其中e的保留时间为2.87min,nmn保留时间为2.74min。
[0113]
步骤1)中,通过外部磁场进行磁性固相萃取仪器转移;在优选的实施例中,采用磁性固相萃取仪器转移磁性固相萃取填料。参照图3,为磁性固相萃取填料转移示意图,其中采用磁性固相萃取仪器为磁棒,磁棒外套有搅拌套,通过磁棒的磁性收集磁珠(磁性固相萃取填料)。磁性固相萃取填料可采用磁性微球或磁珠等弱阳离子交换型磁性固相萃取填料。
[0114]
实施例2对本发明的检测方法进行实验评价
[0115]
1、基质效应评价
[0116]
本实验中通过样本处理后加入法对基质效应进行评价,即mf=提取后的空白基质加入待测物的响应/纯溶剂中待测物的响应。内标归一化mf=待测物mf/内标mf,当内标归一化mf的范围为0.8-1.2可接受。每个待测物选取三个浓度,每个浓度的样本为5份,进行测定。结果如表4-表9所示,表明各化合物的mf均在接受范围内。
[0117]
表4 e的内标归一化
[0118][0119]
表5 ne的内标归一化
[0120][0121]
表6 da的内标归一化
[0122][0123]
表7 mn的内标归一化
[0124]
[0125]
表8 nmn的内标归一化
[0126][0127]
表9 3-mt的内标归一化
[0128][0129]
2、定量下限
[0130]
根据临床检测需求以及仪器设备的灵敏度,将目标物的方法定量下限定为:e为12.5pg/ml、ne为40pg/ml、da为12.5pg/ml、3-mt为5pg/ml、mn为5pg/ml、nmn为12.5pg/ml,连续检测10次,计算检测值和理论值的偏差以及测试结果的变异系数cv值。测试结果如表10所示。由结果可知,测试值和理论值的偏差均在15%以内,测试的平行样本变异系数在20%以内,符合临床检测方法学要求。
[0131]
表10定量下限样本测试结果
[0132][0133]
3、精密度
[0134]
选取低、中、高三个浓度评价方法的精密度,每个批次每个浓度样本分不少于5次进行处理,连续测定3个批次,总样本数不少于45份,分别评估每个浓度样本的批内精密度和批间精密度以及总精密度。结果如表11-表16所示。
[0135]
表11 e的方法精密度
[0136][0137]
[0138]
表12 ne的方法精密度
[0139][0140]
表13 da的方法精密度
[0141][0142][0143]
表14 mn的方法精密度
[0144][0145]
表15 nmn的方法精密度
[0146][0147]
表16 3-mt的方法精密度
[0148]
[0149][0150]
由数据结果可知,低、中、高三个浓度的批内精密度、批间精密度以及总精密度cv均在15%以内,符合方法学验证要求。
[0151]
4、正确度
[0152]
采用真实混合人血清 样,每个样本测试三次,计算加标回收率和检测结果的变异系数。结果如表17-表22所示。由数据结果可知,加标样品的检测值和理论值的偏差符合
±
15%范围内。
[0153]
表17 e的方法回收率
[0154][0155]
表18 ne的方法回收率
[0156][0157]
[0158]
表19 da的方法回收率
[0159][0160]
表20 mn的方法回收率
[0161][0162]
表21 nmn的方法回收率
[0163][0164][0165]
表22 3-mt的方法回收率
[0166][0167]
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。