1.本发明涉及游离氨基酸测定领域,特别涉及一种基于二维色谱分离测定肽类产品中游离氨基酸的方法及其应用。
背景技术:
2.现有的作为食品配料的蛋白肽类产品的国家标准或行业标准(例如gb/t22729-2008)中关于肽含量的测定方法,均规定:肽含量=酸溶蛋白含量-游离氨基酸含量。由于该类产品中主要成分是蛋白质的酶解产物,即相对分子质量大多为10000以下的肽类混合物,另外还含有少量游离氨基酸。按照现有肽类产品的国标或行标要求,此类产品中肽的含量一般至少70%以上,游离氨基酸含量一般大多为5%上下,至多10%~15%。其中,游离氨基酸含量的测定方法是按照相关肽类产品的国标或行标,将样品用5%tca或3.5%磺基水杨酸处理后沉淀去除可能存在的大分子蛋白类成分,再采用以下两种色谱方法之一分离检测游离氨基酸,即方法1:采用gb5009.124-2016的规定的氨基酸分析仪方法,利用阳离子交换色谱法分离,再柱后茚三酮衍生化和紫外检测;方法2:使用柱前opa和fmoc-cl衍生化后再反相色谱法分离,然后再紫外检测或荧光检测。
3.由于肽类样品中肽含量远大于游离氨基酸,故采用上述肽类国标或行标方法,为测定肽含量而采用氨基酸分析仪或柱前衍生化hplc方法检测游离氨基酸含量时,由于肽类同样会发生衍生化反应,游离氨基酸的色谱峰会受到肽类的严重干扰,即两类物质的色谱峰大多会不同程度的重叠,导致游离氨基酸的色谱峰面积积分及其含量计算结果误差较大,加之不同实验室检测人员对重叠峰的切分方式不同,使得不同实验室结果的重现性也较差。因此,提出一种新的可较准确测定肽类产品中游离氨基酸含量的液相色谱测定方法,这对于尚不具备液相色谱-串联质谱分析氨基酸条件的质检机构检测肽类产品具有重要应用价值。
技术实现要素:
4.本发明的首要目的在于克服现有技术的缺点与不足,提供一种基于二维色谱分离测定肽类产品中游离氨基酸的方法。
5.本发明的另一目的在于提供所述基于二维色谱分离测定肽类产品中游离氨基酸的方法的应用。
6.本发明的目的通过下述技术方案实现:
7.一种基于二维色谱分离测定肽类产品中游离氨基酸的方法,包括如下步骤:
8.(1)样品预处理
9.将待测肽类产品用三氟乙酸溶液或磺基水杨酸溶液溶解,离心或过滤后取上清液,得到预处理后的样品溶液;
10.(2)凝胶色谱分离
11.①
硅胶基质的凝胶色谱分离:采用硅胶基质的凝胶柱(凝胶粒)对步骤(1)中得到的预处理后的样品溶液进行色谱分离;其中,色谱分离的条件为:色谱柱:硅胶基质色谱柱;流动相:体积百分比30%~45%的乙腈和体积百分比0.15%~0.25%的三氟乙酸;紫外检测器的检测波长:220nm;流速:0.4~0.6ml/min;柱温:20~35℃;
12.或
13.②
多糖型凝胶色谱分离:采用多糖型凝胶色谱柱对步骤(1)中得到的预处理后的样品溶液进行色谱分离;其中,色谱分离的条件为:色谱柱:多糖型凝胶色谱柱;流动相:50mmol/l磷酸盐缓冲溶液和0.15~0.25mol/l nacl溶液,ph6.5~7.2;紫外检测器的检测波长:220nm:流速:0.4~0.6ml/min;柱温:20~35℃;
14.(3)中心切割
15.根据步骤
①
或
②
中的凝胶色谱分离结果进行流分切割收集,收集起点的相对分子质量为220,并依据最后一个色谱峰出完峰的时间确定结束收集时间,收集的次数为3~5次,合并流分再冷冻干燥,然后用ph 2.2的柠檬酸钠缓冲液溶解定容,膜过滤,得到待测游离氨基酸溶液;
16.(4)第二维色谱分离分析及紫外可见光检测:
17.将步骤(3)中得到的游离氨基酸溶液采用氨基酸自动分析仪或反相hplc色谱进行分离,再采用紫外可见光进行检测,得到待测肽类产品中氨基酸的含量。
18.步骤(1)中所述的肽类产品为食品配料中的所有蛋白肽类产品(蛋白肽类食品配料);包括动物来源的和植物来源的蛋白肽类食品配料;优选为牡蛎肽和大豆肽。
19.步骤(1)中所述的三氟乙酸溶液的浓度为体积百分比5%。
20.步骤(1)中所述的磺基水杨酸溶液的浓度为质量体积比3.5%。
21.步骤(1)中所述的预处理后的样品溶液的浓度为20~25mg/ml;优选为20~23mg/ml。
22.步骤(2)
①
中所述的色谱柱优选为硅胶基质的凝胶柱tsk gel g2000 swxl300mm
×
7.8mm。
23.步骤(2)
①
中所述的流动相优选为:体积百分比40%的乙腈和体积百分比0.1%的三氟乙酸。
24.步骤(2)
①
中所述的流速优选为0.5ml/min。
25.步骤(2)
①
中所述的柱温优选为25℃。
26.步骤(2)
①
中所述的色谱柱优选为多糖型凝胶色谱柱superdex peptide10/300。
27.步骤(2)
②
中所述的流动相优选为:50mmol/l磷酸盐缓冲溶液和0.15mol/lnacl溶液,ph 7.0。
28.步骤(2)
②
中所述的流速优选为0.4ml/min。
29.步骤(2)
②
中所述的柱温优选为30℃。
30.步骤(3)中所述的采用硅胶基质的凝胶色谱分离收集流分的时间为24.5min~30min。
31.步骤(3)中所述的采用多糖型凝胶色谱分离收集流分的时间为43min~54min。
32.步骤(3)中所述的柠檬酸钠缓冲液的用量为0.5~1.0ml。
33.步骤(3)中所述的膜过滤优选为过0.45μm水相滤膜(即0.45μm水相微孔滤膜)。
34.步骤(4)中所述的第二维色谱分离分析旨在将收集得到的游离氨基酸流分,用于后续进一步色谱分离测定。
35.步骤(4)中所述的第二维色谱分离分析及紫外可见光检测优选为通过如下任一种方式实现:
36.(i)将步骤(3)中得到的游离氨基酸溶液利用氨基酸自动分析仪采用阳离子交换色谱分离模式进行分离,并采用柱后茚三酮衍生化,然后进行紫外可见光检测,获得待测肽类产品中氨基酸的含量;其中,紫外可见光检测的条件为:脯氨酸及羟脯氨酸的检测波长为440nm,除脯氨酸和羟脯氨酸以外的氨基酸的检测波长为570nm;
37.(ii)将步骤(3)中得到的游离氨基酸溶液利用hplc自动进样器进行柱前衍生化反应和自动进样,并经反相色谱柱分离后再进行紫外可见光检测,获得待测肽类产品中氨基酸的含量;其中,紫外可见光检测的条件为:脯氨酸及羟脯氨酸的检测波长为262nm,除脯氨酸和羟脯氨酸以外的氨基酸的检测波长为338nm。
38.方式(ii)中所述的柱前衍生化可选用柱前在线自动衍生化或其他柱前衍生化;若是柱前在线自动衍生化,则可选用opa-fmocl衍生化方法,其反应产物经反相色谱分离后进行紫外检测,波长为:338nm(除脯氨酸和羟脯氨酸以外的氨基酸)和262nm(脯氨酸及羟脯氨酸);若采用产物较稳定的其他柱前衍生化反应,经反相色谱分离后,则选用其他相应的波长进行紫外或荧光检测;优选为opa-fmocl衍生化(因可在线自动衍生,检测效率高)。
39.所述的基于二维色谱分离测定肽类产品中游离氨基酸的方法在测定肽类产品中游离氨基酸含量中的应用。
40.所述的肽类产品为食品配料中的所有蛋白肽类产品,即蛋白肽类食品配料;包括动物来源的和植物来源的蛋白肽类食品配料;优选为牡蛎肽和大豆肽。
41.本发明相对于现有技术具有如下的优点及效果:
42.1、本发明方法包括如下几个步骤:样品处理、高效凝胶过滤色谱分离、中心切割(即将凝胶色谱分离流分中游离氨基酸级分切割收集并浓缩后用于后续第二维色谱分离及检测)、第二维色谱分离及衍生化反应,即阳离子交换色谱分离耦合柱后茚三酮衍生化反应(可采用氨基酸自动分析仪进行)或柱前衍生化耦合反相色谱分离、以及紫外(或荧光)检测,该方法通过离线二维色谱分离和中心切割去除蛋白肽类食品配料样品中存在的主要成分即肽类对少量游离氨基酸检测的严重干扰,以达到准确测定游离氨基酸含量的效果。
43.2、由于常规方法测定肽类食品配料中的少量游离氨基酸时,经紫外检测后肽的色谱峰有许多会与氨基酸的色谱峰重叠或部分重叠,因而导致游离氨基酸含量很难准确测定,误差较大,本发明中将hpgfc(高效凝胶色谱法)-hpcec(高效阳离子交换色谱)二维色谱联用,排除肽对游离氨基酸含量测定的影响,利用高效凝胶色谱法将样品中游离氨基酸级分和肽级分基本分离,并采用“中心切割”的方法将游离氨基酸流分切割到第二维色谱即阳离子交换色谱系统中进行分离,同时进行柱后茚三酮衍生化反应和紫外(或荧光)检测各游离氨基酸组分;或将hpgfc(高效凝胶色谱法)-rplc(反相色谱)二维色谱联用,排除肽对游离氨基酸检测的影响,利用高效凝胶色谱法将“中心切割”收集的游离氨基酸级分,利用自动进样器先进行柱前衍生化反应再自动进样至第二维色谱即反相色谱系统,对所有游离氨基酸进行分离,然后在线紫外(或荧光)检测各游离氨基酸组分。
附图说明
44.图1是本发明实施例1中的切割收集氨基酸组分的示意图(蓝色虚线方框表示收集氨基酸组分的区域)。
45.图2是本发明实施例1中利用氨基酸分析仪测定氨基酸收集组分的色谱图。
46.图3是本发明实施例1中利用液相色谱-串联质谱联用仪验证的lc-ms/ms图谱。
47.图4是本发明实施例2中利用多糖型凝胶柱分离收集氨基酸组分的示意图(蓝色虚线方框表示收集氨基酸组分的区域)。
48.图5是本发明实施例2中利用opa/fmoc-cl柱前衍生化反相色谱测定大豆肽样品经多糖型凝胶色谱柱分离收集的氨基酸组分色谱图。
49.图6是本发明实施例1中利用液相色谱-串联质谱联用仪验证的lc-ms/ms图谱。
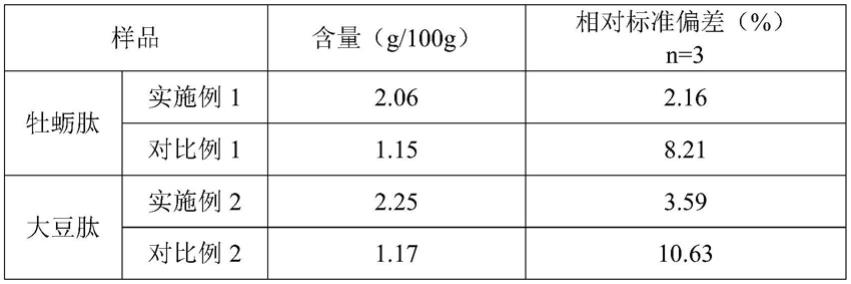
50.图7是本发明对比例1中的样品经5%tca处理后上清液直接色谱分析图。
51.图8是本发明对比例2中的样品经3.5%磺基水扬酸处理后上清液直接色谱分析图。
具体实施方式
52.下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。下列实施例中未注明具体实验条件的试验方法,通常按照常规实验条件或按照制造厂所建议的实验条件。除非特别说明,本发明所用试剂和原材料均可通过市售获得。
53.实施例1
54.本实施例提供一种基于二维色谱分离测定牡蛎肽产品中游离氨基酸的方法,具体包括如下步骤:
55.(1)样品预处理:准确称取200.0mg的牡蛎肽粉(市售产品),溶于10ml的5%(v/v)三氟乙酸溶液,过滤后取上清液,在高效凝胶色谱系统上进样和分离制备。
56.(2)凝胶色谱分离:采用硅胶基质的凝胶柱tsk gel g2000 swxl 300mm
×
7.8mm,流动相是:40%(v/v)乙腈(acn)/0.1%(v/v)三氟乙酸(tfa),其监测所用紫外检测器的检测波长为220nm;流速为0.5ml/min;柱温:25℃。
57.(3)中心切割:通过凝胶色谱分离去除绝大多数肽组分,收集游离氨基酸流分,凝胶色谱分离的流分切割收集起点的相对分子质量为220(在相对分子质量最大的氨基酸的保留时间适当提前收集,蛋白质组成中相对分子质量最大的氨基酸是色氨酸,其相对分子质量为204.22)。为确定其对应的保留时间,便于准确收集流分,首先利用不同相对分子质量的肽类标品在上述凝胶色谱分离条件下制作相对分子质量-保留时间校正曲线及其方程(校正曲线方程:lgmw=7.01-0.192t;其中,mw为相对分子质量,t为保留时间),其系列标品为:(i)细胞色素c,mw 12384;(ii)抑肽酶,mw 6512;(iii)杆菌酶,mw 1423:(iv)乙氨酸-乙氨酸-酪氨酸-精氨酸,mw 451;(v)乙氨酸-乙氨酸-乙氨酸,mw 189(系列标品均购自sigma公司)。根据校正曲线方程计算得到的相对分子质量220所对应的保留时间确定收集流分的起始时间为24.5min,依据最后一个色谱峰出完峰的时间确定结束收集时间(本实验的结束收集时间为30min),收集4次,合并流分再冷冻干燥,然后用0.5ml的ph 2.2柠檬酸钠缓冲液溶解定容(参考gb 5009.124-126),过0.45微米的水相微孔滤膜后转移至进样瓶中进行氨
基酸分析。
58.(4)第二维色谱分离分析:将收集得到的游离氨基酸流分,上氨基酸自动分析仪进行分析测定,其分离是使用阳离子交换色谱柱,并采用柱后茚三酮衍生化(即按照gb 5009.124-2016所述方法进行),再进行后续紫外可见光检测。
59.(5)紫外可见光检测:可见光分光光度的检测波长为570nm(除脯氨酸和羟脯氨酸以外的氨基酸)和440nm(检测脯氨酸和羟脯氨酸)。
60.图1为牡蛎肽样品的相对分子质量分布色谱图及按照上述步骤切割收集游离氨基酸组分的虚线方框示意图,图2为氨基酸分析仪测定氨基酸收集组分的色谱图:经氨基酸分析仪分析和计算得到上述牡蛎肽样品中的游离氨基酸含量为2.06g/100g。
61.(6)验证:采用液相色谱-串联质谱联用仪(lc-ms/ms)进行验证(方法依据游离氨基酸的测定标准:gb/t 30987-2020),测定得到的牡蛎肽样品中的游离氨基酸含量是2.15g/100g(lc-ms/ms图谱见图3),本发明测定方法与液相色谱-串联质谱联用仪测定的结果接近,其相对误差为4.27%,在允许范围内。
62.实施例2
63.本实施例提供一种基于二维色谱分离测定大豆肽样品中游离氨基酸的方法,具体操作方法如下:
64.(1)样品预处理:准确称量230.0mg大豆肽样品(市售产品),用3.5%(w/v)磺基水杨酸溶液溶解定容至10ml,即浓度为23.0mg/ml,离心并过滤后在高效凝胶色谱系统上进样和分离制备。
65.(2)凝胶色谱分离:采用多糖型凝胶色谱柱superdex peptide 10/300,流动相是:50mmol/l磷酸盐缓冲溶液 0.15mol/l nacl溶液,ph 7.0,紫外检测器的检测波长为220nm:流速均为0.4ml/min;柱温为:30℃。
66.(3)中心切割:凝胶色谱分离的流分切割收集起点的相对分子质量为220左右。为确定其对应的保留时间,便于准确收集流分,也首先利用不同相对分子质量的肽类标品在上述凝胶色谱分离条件下制作相对分子质量-保留时间校正曲线及其方程,其系列标品与实施例1相同。收集流分的开始时间点由校正曲线方程计算得到的相对分子质量220所对应的保留时间确定,依据最后一个色谱峰出完峰的时间确定结束收集时间(本实验收集流分的起始时间为43min,结束收集时间为54min)收集3次,合并流分再冷冻干燥后,用1ml的ph 2.2柠檬酸钠缓冲液溶解定容(参考gb 5009.124-126),过0.45微米的水相微孔滤膜后转移至进样瓶中进行后续氨基酸分析。
67.(4)第二维色谱分离:选用反相色谱模式,采用反相hplc色谱柱(hypersil ods 4.0mm
×
250mm,5μm)分离,利用hplc自动进样器进行柱前衍生化反应和自动进样(采用opa/fmoc-cl衍生化反应,其反应速度较快且反应产物不够稳定,需尽快进样检测,其具体色谱条件参照gb/t 22729-2008中6.3.2.2.2.4所述色谱条件进行;若选用其他的柱前衍生化方法且产物较稳定,其柱前衍生化反应也可手工操作)。
68.(5)紫外检测:采用柱前在线自动衍生化(opa-fmocl衍生化方法),反相色谱分离后的紫外检测波长为:262nm(脯氨酸及羟脯氨酸)和338nm(除脯氨酸和羟脯氨酸以外的其他氨基酸)。
69.图4为大豆肽样品经多糖型凝胶柱分离分析的相对分子质量分布色谱图及按照上
述步骤切割收集游离氨基酸组分的虚线方框示意图。经opa/fmoc-cl柱前衍生化反相色谱分析(图5)和计算得到其大豆肽样品中游离氨基酸含量为2.25g/100g。
70.(6)验证:采用液相色谱-串联质谱联用仪进行验证(方法依据gb/t30987-2020),测定得到的大豆肽样品中的游离氨基酸含量是2.39g/100g(lc-ms/ms图谱见图6)。本发明测定方法与液相色谱-串联质谱联用仪测定的结果接近,其相对误差为6.03%,在允许范围内。
71.对比例1
72.按实施例1中的方法,对牡蛎肽粉样品经5%(v/v)tca预处理后的上清液直接进行色谱分离(实施例1步骤(4)),其色谱分析结果见图7。
73.对比例2
74.按实施例2中的方法,对大豆肽样品经3.5%(w/v)磺基水扬酸预处理后的上清液直接进行色谱分析(实施例2步骤(4)),其色谱分析结果见图8。
75.效果实施例
76.本发明实施例1-2以及对比例1-2的比较结果如表1所示(均为三次重复测定的结果)。
77.表1本发明与对比例方法的比较
[0078][0079]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。