1.本发明属于信息处理领域,具体涉及一种基因测序多样本数据文件自动配对的方法和系统。
背景技术:
2.在基因测序过程中,单端测序是最简单的测序方法,采用单一的测序引物,使得pcr沿着该测序引物的方向进行,因此所有测序片段(reads)只能按照一个方向读取,但测序的质量会随着测序进行而下降,导致越往后测序,reads越不准确。为了克服这一缺点,双末端测序技术开始普及,从两端向中间进行两个不同方向的测序,获得两个方向的reads,每一个read的长度都要超过整个待测序列的一半,再根据匹配的两个reads重合的部分进行拼接计算,得到最终确认的基因序列,不但可以大大提高测序准确率,而且还有利于检测出单端测序数据无法检出的插入缺失变异。
3.随着基因测序技术的不断发展,第二代测序(next-generation sequencing,ngs)作为一种基于pcr和基因芯片发展而来的dna测序技术,具有测序通量高、速度快、成本低的优势,取得了越来越广泛的应用。然而,高通量测序检测的样本量非常庞大,因此双通量测序方法会获得大量记载reads信息的文件,要进行后续的基因数据计算,必须将记载了对同一样本测序得到的两个方向的reads信息的一对数据文件(模板链文件和互补链文件)进行匹配,因此,在多样本的基因测序数据文件中如何快速准确地配对每个样本的模板链和互补链文件是一大难题。
4.现有的配对方法多是通过测序生成的样本的文件名称来进行模板链文件和互补链文件匹配,以及模板链和互补链的区分,然而,大量测序样本的数据文件仅通过文件名称难以管理,且文件名称很容易被人为修改,最终导致配对程序无法正常执行。
5.因此,随着测序样本越来越多,针对以上难题,提供一种能够快速准确地配对样本数据文件,并区分同一样本模板链文件和互补链文件的技术具有重要意义。
技术实现要素:
6.本发明的目的在于提供一种基因测序多样本数据文件自动配对的方法和系统。
7.本发明提供了一种基因测序多样本数据文件自动配对的方法,包括如下步骤:(1)读取待配对基因测序多样本数据文件的内容;(2)获取测序片段id的信息,分别将每个基因测序多样本数据文件的测序片段id信息保存为对应的临时文件;(3)采用信息摘要算法计算得到各临时文件的摘要;(4)对比各临时文件的摘要,摘要一致的即为配对的基因测序多样本数据文件;所述基因测序多样本数据文件为符合国家标准gb/t 35890-2018的fastq格式的文件。
8.进一步地,步骤(1)所述读取为按行读取fastq文件的内容,并去除行首和行尾的
特殊字符;所述特殊字符包括空格字符、回车字符。
9.进一步地,步骤(2)所述获取测序片段id的信息是获取文件中以字符@开头的一行信息。
10.进一步地,步骤(3)所述信息摘要算法是md5算法。
11.更进一步地,步骤(1)所述待配对基因测序多样本数据文件是通过如下步骤获取的:(a)查找确认存放目录下双末端测序得到的所有基因测序多样本数据文件;(b)判断文件查找模块确认的基因测序多样本数据文件是否是压缩文件,对压缩文件解压;优选地,步骤(a)所述查找确认的方法是:解析文件名称,文件名称后缀字符包含fq或fastq即确认进行步骤(b);和/或步骤(b)所述判断是否是压缩文件的方法是:解析文件名称,文件名称后缀字符为gz、zip或bz的文件为压缩文件。
12.进一步地,上述方法还包括区分配对的2个基因测序多样本数据文件中模板链文件和互补链文件的步骤:比较配对的2个基因测序多样本数据文件中测序片段id信息中的标识信息大小,数值小的是模板链文件,数值大的是互补链文件。
13.本发明还提供了一种基因测序多样本数据文件自动配对的系统,它包括按照上述的方法对基因测序多样本数据文件进行配对的模块。
14.进一步地,它包括如下模块:文件读取模块:读取带配对基因测序多样本数据文件的内容;文件解析模块:获取测序片段id的信息,分别将每个基因测序多样本数据文件的测序片段id信息保存为对应的临时文件;信息摘要算法模块:采用信息摘要算法计算得到各临时文件的摘要;配对模块:对比各临时文件的摘要,摘要一致的确定为配对的基因测序多样本数据文件。
15.更进一步地,它还包括如下模块:文件查找模块:查找确认存放目录下双末端测序得到的所有需要处理的基因测序多样本数据文件;文件解压模块:判断文件查找模块确认的文件是否是压缩文件,对压缩文件解压;优选地,它还包括异常处理模块:记录其它模块处理过程中出现的异常信息;和/或它还包括区分模块:比较配对的2个基因测序多样本数据文件中测序片段id信息中的标识信息大小,数值小的是模板链文件,数值大的是互补链文件。
16.本发明还提供了一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序用于实现上述的基因测序多样本数据文件自动配对的方法。
17.基因数据具有海量性,本发明方法可以方便管理大量基因测序数据文件,减少因人为造成的程序执行错误问题,提高计算机资源的使用效率。
18.除另有说明外,本发明涉及的术语与国家标准gb/t 35890-2018中定义的术语和描述规范的含义一致。
19.本发明所述的“测序片段id信息中的标识信息”,是指通过双端测序直接自动生成
的fastq文件中的测序片段id行的标识信息,例如对于illumina测序得到的fastq文件而言,就是以字符“/”(早期数据)或者是字符空格
“ꢀ”
(后期数据)后的数值大小来标识模板链或互补链。
20.显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
21.以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
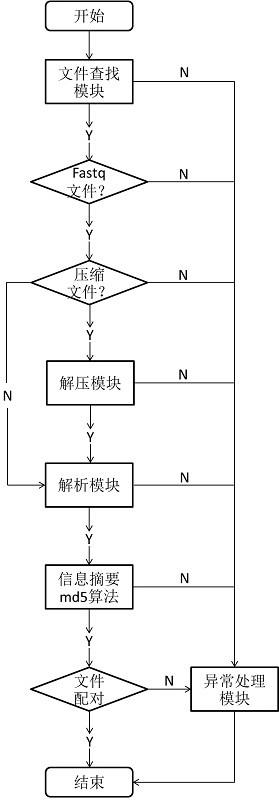
22.图1为本发明方法对文件进行配对处理的流程图。
具体实施方式
23.实施例1、本发明方法配对基因测序多样本数据文件1)文件查找模块判读是否是文件,然后遍历所有文件,解析文件名称,文件名称后缀字符包含fq和fastq,就是需要处理的fastq文件,其他文件不会处理。
24.2)文件解压模块判断文件名称后缀为.gz、.zip和.bz的文件为压缩文件,需要进行解压处理,解压成文本文件,其他文件不处理。处理过程中,如果出现异常由异常处理模块处理。
25.3)文件读取模块按行读取fastq文件内容,去除行首和行尾的空格和回车等特殊字符。处理过程中,如果出现异常由异常处理模块处理。
26.4)文件解析模块按照《高通量测序数据序列格式规范》(国家标准号:gb/t 35890-2018)6.1节“fastq格式”中规定的规范解析文件,首字符@开头的行为测序片段的信息行,每个数据文件只需要存储测序片段的信息行。处理过程中,如果出现异常由异常处理模块处理。
27.5) 信息摘要算法模块用md5算法对每个临时中间文件存储的测序片段信息行生成摘要。摘要信息一致的就是配对的样本,配对的样本应该只有一对,即两个文件。如果配对的文件不是两个(一个文件或多个文件),则交给异常处理模块处理。
28.6)区分模块将配对成功的样本,根据测序片段id的标识信息大小就可以区分出模板链文件及互补链文件(数值小的是模板链,数值大的是模板链)。
29.7)异常处理模块收到异常后,不会中断执行,只是记录异常信息,记录的信息包括,异常样本的文件名,文件解压错误、文件读取错误、文件解析错误,没有配对的样本(一个样本文件或多个样本文件)。
30.8)使用方法:执行程序的时候,参数必须输入fastq文件存放的目录。程序会根据目录找出所有样本,生成一个配对成功的文件(每行一个配对的样本)。其他异常情况,会生成一个异常错误的文件,供用户查看并处理异常的样本。图1为上述方法对文件进行配对处理的流程图。
31.以下通过实验例证明本发明的有益效果。
32.实验例1、本发明方法的自动配对效果
在/fastq1目录下,存放有1450个样本的2900个fastq数据文件,传统方式是由人工通过文件名称整理每个样本的配对数据文件至少需要1个小时。
33.使用本发明实施例1自动配对的方法系统可以快速将文件进行匹配输出,并作出模板链和互补链的区分标识,两个配对数据文件的文件名前缀相同,后缀包含r1字符的为模板链数据文件,后缀包含r2字符的为互补链数据文件。整个过程仅需要不到3秒的时间,同时还有效发现两个错误,分别为数据文件匹配不上和文件解压的错误。
34.可见,本发明方法可以准确、有效、快速地对大量基因测序多样本数据文件进行配对处理,大大缩短人工整理的时间成本,具有推广应用价值。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。