1.本发明属于有机物检测技术领域,具体涉及一种米糠脂肪烷醇中二十八碳脂肪烷醇和三十碳脂肪烷醇含量的检测方法。
背景技术:
2.米糠脂肪烷醇是以米糠为原料制得米糠蜡后经皂化、提取、过滤等工艺制成的十二个碳原子以上蜡状固体的饱和一元醇,其主要成分包括二十八碳脂肪烷醇、三十碳脂肪烷醇、三十二碳脂肪烷醇等高级脂肪烷醇。米糠脂肪烷醇的含量一般以二十八碳脂肪烷醇、三十碳脂肪烷醇含量之和计。米糠脂肪烷醇作为一种天然的新食品添加剂,已经被证实在改善身体状态以及保健等多方面具有良好的功效。米糠脂肪烷醇中二十八碳脂肪烷醇具有增进体力、增强耐力和精力、提高反应灵敏性和应激能力、提高机体代谢率、改善心肌功能、降低血清胆固醇和甘油三酯含量、降低收缩期血压等功能,可以作为一种新型功能性食品添加剂和营养补助品等健康食品。三十碳脂肪烷醇具有促进生根、发芽、开花、茎叶生长和早熟作用,具有提高叶绿素含量、增强光合作用等多种生理功能,因为也被广泛应用于农作物上。米糠脂肪烷醇的研究开发不仅能为人们提供多种用于医药、保健、化妆品、农作物等方面的产品,而且还能变废为宝,合理利用资源,保护环境的同时,让米糠油的重要副产品米糠蜡利益最大化。
3.目前,国内还未发布高级脂肪烷醇在米糠脂肪烷醇中的检测标准,由于没有该产品的国家级或省级检测方法,检测机构没有资质为企业出具米糠脂肪烷醇中高级脂肪烷醇含量的检测报告,造成生产企业无法提供米糠脂肪烷醇的全检报告来申报食品生产许可证(sc证)。目前,检测有机化合物的方法有红外光谱法、紫外光谱法和气相色谱法等,但根据高级脂肪烷醇的特性发现,红外光谱虽然可以表征羟基(-oh)、末端甲基(-ch3)、次甲基(-ch2)以及(-ch
2-)n特有的吸收峰,证明米糠脂肪烷醇含有饱和碳直链的伯醇,但二十八碳脂肪烷醇和三十碳脂肪烷醇是同系物,都有类似的吸收峰,因而不能准确对其进行定性和定量,另外研究人员对二十八碳脂肪烷醇和三十碳脂肪烷醇标样进行紫外光谱扫描时发现,二十八碳脂肪烷醇和三十碳脂肪烷醇在紫外光谱中的特征吸收波长分别在198nm和200nm处,非常接近,因而采用紫外检测器对其定量也不可行。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种米糠脂肪烷醇中二十八碳脂肪烷醇和三十碳脂肪烷醇含量的检测方法,本发明提供的检测方法具有高精确度,二十八碳脂肪烷醇的检出限为0.07%,定量限为0.20%,三十碳脂肪烷醇的检出限为0.08%,定量限为0.25%。
5.为了实现上述目的,本发明提供了以下技术方案:
6.本发明提供了一种米糠脂肪烷醇中二十八碳脂肪烷醇和三十碳脂肪烷醇含量的检测方法,包括以下步骤:
7.将待测样品和有机溶剂混合,进行超声提取,得到待测样品液;
8.采用气相色谱法对所述待测样品液进行检测,得到二十八碳脂肪烷醇和三十碳脂肪烷醇的含量;
9.所述气相色谱法中柱温箱的升温程序为室温180~220℃,保持1min,然后以10~15℃/min的速率升温至260~300℃,保持20min。
10.优选的,所述有机溶剂包括乙醇、二氯甲烷、三氯甲烷、甲苯、环己烷、正己烷、异丙醇、石油醚、甲醇或丙酮。
11.优选的,所述待测样品和有机溶剂的用量比为(0.02~0.2)g∶100ml。
12.优选的,所述超声提取的温度为20~50℃。
13.优选的,所述超声提取的时间为5~30min。
14.优选的,所述超声提取的功率为300~500w。
15.优选的,所述气相色谱法所用色谱柱为安捷伦中等极性db-1701或安捷伦弱极性hp-5色谱柱。
16.优选的,所述气相色谱法的进样模式为不分流进样或分流进样;当所述气相色谱法的进样模式为分流进样时,分流比为5∶1。
17.优选的,所述气相色谱法的进样口温度为300℃;所述气相色谱法的检测器温度为320℃。
18.优选的,所述气相色谱法的进样量为2μl;所述气相色谱法的载气流速为1~3ml/min。
19.本发明提供了一种米糠脂肪烷醇中二十八碳脂肪烷醇和三十碳脂肪烷醇含量的检测方法,包括以下步骤:将待测样品和有机溶剂混合,进行超声提取,得到待测样品液;采用气相色谱法对所述待测样品液进行检测,得到二十八碳脂肪烷醇和三十碳脂肪烷醇的含量;所述气相色谱法中柱温箱的升温程序为室温180~220℃,保持1min,然后以10~15℃/min的速率升温至260~300℃,保持20min。本发明中采用超声提取的方法对待测样品进行提取,超声提取具有提取温度低、提取率高、提取时间短的特点,能够有效提取待测样品中的二十八碳脂肪烷醇和三十碳脂肪烷醇,为后续定量检测奠定基础,采用气相色谱法测定米糠脂肪烷醇中二十八碳脂肪烷醇和三十碳脂肪烷醇,并通过优化升温程序,提高二十八碳脂肪烷醇和三十碳脂肪烷醇的分离度,优化组分峰形,实现对米糠脂肪烷醇中二十八碳脂肪烷醇和三十碳脂肪烷醇的快速定性定量分析,具有简便、快捷、精确度高的优点。由实施例结果可知,本发明提供的检测方法确定二十八碳脂肪烷醇的检出限为0.07%,定量限为0.20%,三十碳脂肪烷醇的检出限为0.08%,定量限为0.25%。本发明的检测方法补充米糠脂肪烷醇的检测,为检测米糠脂肪烷醇提供技术支撑,弥补了目前针对米糠脂肪烷醇检测方法的空白,值得推广应用。
附图说明
20.图1为外标法中二十八碳脂肪烷醇和三十碳脂肪烷醇的标准曲线图;
21.图2为内标法中二十八碳脂肪烷醇和三十碳脂肪烷醇的标准曲线图;
22.图3为实施例2~11中有机溶剂提取二十八碳脂肪烷醇和三十碳脂肪烷醇的响应情况图;
23.图4为不同超声时间下二十八碳脂肪烷醇的提取率图;
24.图5为不同超声时间下三十碳脂肪烷醇的提取率如图;
25.图6为不同超声温度下二十八碳脂肪烷醇的提取率图;
26.图7为不同超声温度下三十碳脂肪烷醇的提取率图;
27.图8为不同超声功率下二十八碳脂肪烷醇的提取率图;
28.图9为不同超声功率下三十碳脂肪烷醇的提取率图;
29.图10为苯甲酸苯酯的色谱图;
30.图11为邻苯二甲酸二癸酯的色谱图;
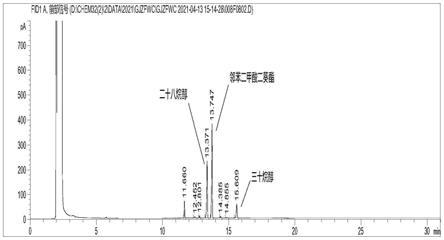
31.图12为在米糠脂肪烷醇样品添加邻苯二甲酸二癸酯的色谱图;
32.图13为二十八碳脂肪烷醇和三十碳脂肪烷醇在中等极性db-1701(30m
×
0.320mm
×
0.25μm)色谱柱中的色谱图;
33.图14为二十八碳脂肪烷醇和三十碳脂肪烷醇在弱极性hp-5(30m
×
0.320mm
×
0.25μm)色谱柱中的色谱图;
34.图15为进样量2μl,进样口温度300℃,检测器fid温度320℃,流速为3ml/min,进样模式为分流,分流比5:1,分流流量15ml/min,升温程序为起始温度200℃,保持1min,然后以10℃/min的速率升到300℃,保持20min时,二十八碳脂肪烷醇和三十碳脂肪烷醇的色谱图;
35.图16为进样量2μl,进样口温度300℃,检测器fid温度320℃,流速为3ml/min,进样模式为不分流,升温程序为起始温度200℃,保持1min,然后以10℃/min的速率升到300℃,保持20min时,二十八碳脂肪烷醇和三十碳脂肪烷醇的色谱图;
36.图17为进样量2μl,进样口温度300℃,检测器fid温度320℃,流速为3ml/min,进样模式为不分流,升温程序为起始温度200℃,保持1min,然后以10℃/min的速率升到300℃,保持20min时,二十八碳脂肪烷醇和三十碳脂肪烷醇的色谱图。
具体实施方式
37.本发明提供了一种米糠脂肪烷醇中二十八碳脂肪烷醇和三十碳脂肪烷醇含量的检测方法,包括以下步骤:
38.将待测样品和有机溶剂混合,进行超声提取,得到待测样品液;
39.采用气相色谱法对所述待测样品液进行检测,得到二十八碳脂肪烷醇和三十碳脂肪烷醇的含量;
40.所述气相色谱法的进样模式为不分流进样或分流进样;所述气相色谱法中柱温箱的升温程序为室温180~220℃,保持1min,然后以10~15℃/min的速率升温至260~300℃,保持20min。
41.如无特殊说明,本发明对所用原料的来源没有特殊要求,采用本领域技术人员所熟知的市售商品即可。
42.本发明将待测样品和有机溶剂混合,进行超声提取。
43.在本发明中,所述有机溶剂优选包括无水乙醇、二氯甲烷、三氯甲烷、甲苯、环己烷、正己烷、异丙醇、石油醚、甲醇或丙酮,更优选为甲苯、环己烷、正己烷或石油醚;所述石油醚优选为沸点为60~90℃的石油醚;所述待测样品和有机溶剂的用量比优选为(0.02~0.2)g∶100ml,更优选为(0.02~0.05)g∶100ml。本发明对所述待测样品没有特殊限定,采用
本领域熟知的样品即可,在本发明实施例中,所述待测样品具体由浙江省金华市武义县震威生物科技有限公司提供。
44.在本发明中,所述超声提取的温度优选为20~50℃,更优选为30~50℃;所述超声提取的时间优选为5~30min,更优选为15~30min;所述超声提取的功率优选为300~500w,更优选为400~500w;所述超声提取的设备优选为超声波清洗器,在本发明实施例中,所述超声提取的设备具体为2500th数控超声波清洗器。
45.本发明中采用超声提取的方法对待测样品进行提取,超声提取具有提取温度低、提取率高、提取时间短的特点,能够有效提取待测样品中的二十八碳脂肪烷醇和三十碳脂肪烷醇,为后续定量检测奠定基础。
46.超声提取完成后,本发明优选将所述超声提取所得溶液进行静置至室温。
47.静置至室温后,本发明优选对所述静置至室温后的溶液进行定容,得到待测样品液。
48.本发明对所述定容的方法没有特殊限定,采用本领域熟知的定容方法即可。
49.得到待测样品液后,本发明采用气相色谱法对所述待测样品液进行检测,得到二十八碳脂肪烷醇和三十碳脂肪烷醇的含量。
50.在本发明中,所述气相色谱法的进样口温度优选为300℃;所述气相色谱法的检测器温度优选为320℃;所述气相色谱法的进样量优选为2μl;所述气相色谱法的载气流速优选为1~3ml/min,更优选为1~2ml/min;所述气相色谱法的进样模式优选为不分流进样或分流进样;当所述气相色谱法的进样模式为分流进样时,所述分流比优选为5:1;所述气相色谱法中柱温箱的升温程序为室温180~220℃,保持1min,然后以10~15℃/min的速率升温至260~300℃,保持20min,更优选为室温200℃,保持1min,然后以15℃/min的速率升温至260℃,保持20min或室温200℃,保持1min,然后以10℃/min的速率升温至300℃,保持20min,最优选为升温至200℃,保持1min,然后以10℃/min的速率升温至300℃,保持20min。
51.本发明对所述气相色谱法所用仪器没有特殊限定,采用本领域熟知的气相色谱仪即可。在本发明实施例中,所述气相色谱法所用的仪器具体为配火焰离子化检测器(fid)的安捷伦7890a气相色谱仪。
52.在本发明中,所述气相色谱法所用色谱柱优选为安捷伦中等极性db-1701色谱柱或安捷伦弱极性hp-5色谱柱,更优选为安捷伦弱极性hp-5色谱柱。所述安捷伦中等极性db-1701色谱柱的填料为(14%-氰丙基-苯基)-甲基聚硅氧烷,所述安捷伦中等极性db-1701色谱柱的规格为30m
×
0.320mm
×
0.25μm;所述安捷伦弱极性hp-5色谱柱的填料为(5%-苯基)-甲基聚硅氧烷,所述安捷伦弱极性hp-5色谱柱的规格为30m
×
0.320mm
×
0.25μm。
53.本发明优选采用内标法或外标法对所述待测样品中二十八碳脂肪烷醇和三十碳脂肪烷醇进行定量。在本发明中,所述内标法优选为将所述待测样品液和内标液混合后,采用气相色谱法对所述混合液进行检测。在本发明中,所述内标液优选为邻苯二甲酸二癸酯标准液或苯甲酸苯酯标准液,更优选为邻苯二甲酸二癸酯标准液。本发明对所述待测样品液和内标液混合的过程没有特殊限定,采用本领域熟知的混合过程即可。
54.在本发明中,所述内标液的配制过程优选为称取50mg苯甲酸苯酯标准品或邻苯二甲酸二癸酯标准品,用所述有机溶剂配制成5mg/ml的内标液,冷藏保存。
55.在本发明中,所述内标法以二十八碳脂肪烷醇或三十碳脂肪烷醇的保留时间定
性,通过二十八碳脂肪烷醇或三十碳脂肪烷醇与内标物色谱峰面积的比值,根据标准曲线得到二十八碳脂肪烷醇或三十碳脂肪烷醇的含量。
56.在本发明中,所述外标法以二十八碳脂肪烷醇或三十碳脂肪烷醇的保留时间定性,通过二十八碳脂肪烷醇或三十碳脂肪烷醇与同样检测条件下标准物质的色谱峰面积的比值,根据标准曲线得到二十八碳脂肪烷醇或三十碳脂肪烷醇的含量。
57.在本发明中,所述标准曲线的绘制过程具体为:
58.二十八碳脂肪烷醇或三十碳脂肪烷醇标准储备液的配制:准确称取20mg二十八碳脂肪烷醇或三十碳脂肪烷醇标准品(精确至0.01mg)于100ml容量瓶中,用甲苯配制成质量浓度为0.2mg/ml的标准储备液,常温密封保存,备用;
59.二十八碳脂肪烷醇或三十碳脂肪烷醇标准工作溶液的配制:吸取0.5、1.0、2.5、5.0、10.0ml二十八碳脂肪烷醇或三十碳脂肪烷醇标准储备液于10ml容量瓶中,用甲苯定容,配制成0.01、0.02、0.05、0.1、0.2mg/ml的标准工作溶液,常温密封保存;
60.混合脂肪烷醇标准工作溶液的配制:分别吸取0.5、1.0、2.5、5.0、10.0ml二十八碳脂肪烷醇和三十碳脂肪烷醇标准储备液于10ml容量瓶中,用甲苯定容,配制成0.01、0.02、0.05、0.1、0.2mg/ml的混合标准工作溶液,常温密封保存;
61.邻苯二甲酸二癸酯工作液:准确称取50mg邻苯二甲酸二癸酯标准溶液,用甲苯配制成5mg/ml的内标工作液,冷藏保存;
62.外标法:分别吸取1ml脂肪烷醇系列标准工作液,测定目标组分的色谱峰面积,以目标组分峰面积(y)为纵坐标,以其对应的质量浓度(x,mg/ml)为横坐标,作线性回归分析,如表1所示,标准曲线如图1所示;
63.内标法:分别吸取1ml二十八碳脂肪烷醇或三十碳脂肪烷醇标准工作液,加入40μl邻苯二甲酸二癸酯内标工作液,混匀,测定目标峰和内标峰的色谱峰面积,以目标组分跟内标物的峰面积比(y)为纵坐标,以系列浓度(x,mg/ml)为横坐标,作线性回归分析,如表1所示,标准曲线如图2所示。
64.表1二十八碳脂肪烷醇和三十碳脂肪烷醇的线性范围、线性方程和相关系数
[0065][0066]
结果计算:
[0067]
试样中二十八碳脂肪烷醇或三十碳脂肪烷醇含量按式(1)计算:
[0068][0069]
式(1)中:
[0070]
x——试样中二十八碳脂肪烷醇或三十碳脂肪烷醇的质量百分比含量,%;
[0071]
c——从标准曲线中得到的被测组分的浓度,单位为毫克每毫升(mg/ml);
[0072]
v——试样溶液定容体积,单位为毫升(ml);
[0073]
m——试样的质量,单位为克(g)。
[0074]
测定结果取两次测定的算术平均值,结果保留3位有效数字。
[0075]
试样中高级脂肪烷醇的含量(二十八碳脂肪烷醇和三十碳脂肪烷醇)按式(2)计算:
[0076]
x=∑xi
ꢀꢀ
(2)
[0077]
式(2)中:
[0078]
x——试样中高级脂肪烷醇(二十八碳脂肪烷醇和三十碳脂肪烷醇)的质量百分比含量,%;
[0079]
xi——试样中二十八碳脂肪烷醇或三十碳脂肪烷醇的质量百分比含量,%;结果保留3位有效数字。
[0080]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
[0081]
实施例1
[0082]
称取20mg米糠脂肪烷醇样品置于100ml容量瓶中,加入80ml甲苯,在30℃下以500w超声提取5min,使其完全溶解,静置至室温后用甲苯定容至100ml,取1ml滤液,加入40μl内标液(5mg/ml的邻苯二甲酸二癸酯-甲苯溶液),混匀,用气相色谱仪进行测定,色谱柱为安捷伦弱极性hp-5(5%-苯基)-甲基聚硅氧烷(30m
×
0.320mm
×
0.25μm)色谱柱,柱温箱的程序升温为室温200℃,保存1min,以10℃/min的速率升温至300℃,保存20min,进样口温度为300℃,检测器(fid)温度为320℃,进样模式为不分流进样,进样量为2μl,流速为1ml/min。
[0083]
实施例2
[0084]
称取0.05g米糠脂肪烷醇样品用100ml甲苯溶解,取1ml滤液,用气相色谱仪进行测定,色谱柱为安捷伦弱极性hp-5(5%-苯基)-甲基聚硅氧烷(30m
×
0.320mm
×
0.25μm)色谱柱,柱温箱的程序升温为室温200℃,保存1min,以10℃/min的速率升温至300℃,保存20min,进样口温度为300℃,检测器(fid)温度为320℃,进样模式为不分流进样,进样量为2μl,流速为1ml/min。
[0085]
实施例3
[0086]
与实施例2的区别在于,有机溶剂替换为无水乙醇,其余内容与实施例2一致。
[0087]
实施例4
[0088]
与实施例2的区别在于,有机溶剂替换为二氯甲烷,其余内容与实施例2一致。
[0089]
实施例5
[0090]
与实施例2的区别在于,有机溶剂替换为三氯甲烷,其余内容与实施例2一致。
[0091]
实施例6
[0092]
与实施例2的区别在于,有机溶剂替换为环己烷,其余内容与实施例2一致。
[0093]
实施例7
[0094]
与实施例2的区别在于,有机溶剂替换为正己烷,其余内容与实施例2一致。
[0095]
实施例8
[0096]
与实施例2的区别在于,有机溶剂替换为异丙醇,其余内容与实施例2一致。
[0097]
实施例9
[0098]
与实施例2的区别在于,有机溶剂替换为沸点为60~90℃的石油醚,其余内容与实施例2一致。
[0099]
实施例10
[0100]
与实施例2的区别在于,有机溶剂替换为甲醇,其余内容与实施例2一致。
[0101]
实施例11
[0102]
与实施例2的区别在于,有机溶剂替换为丙酮,其余内容与实施例2一致。
[0103]
实施例12
[0104]
称取0.02g含量为各自含量10%、30%、50%、70%和90%的二十八碳脂肪烷醇和三十碳脂肪烷醇的米糠脂肪烷醇样品于100ml容量瓶中,用甲苯溶解,依次改变超声的时间(5,10,15,20,30min),超声的温度(20,25,30,40,50℃),超声的功率(300,350,400,450,500w),取1ml滤液,用气相色谱仪进行测定,色谱柱为安捷伦弱极性hp-5(5%-苯基)-甲基聚硅氧烷(30m
×
0.320mm
×
0.25μm)色谱柱,柱温箱的程序升温为室温200℃,保存1min,以10℃/min的速率升温至300℃,保存20min,进样口温度为300℃,检测器(fid)温度为320℃,进样模式为不分流进样,进样量为2μl,流速为1ml/min。
[0105]
实施例13
[0106]
与实施例1的区别在于,内标液为5mg/ml的苯甲酸苯酯-甲苯溶液,其与内容与实施例1一致。
[0107]
测试结果:
[0108]
(1)标准曲线的绘制:
[0109]
二十八碳脂肪烷醇或三十碳脂肪烷醇标准储备液的配制:准确称取20mg二十八碳脂肪烷醇或三十碳脂肪烷醇标准品(精确至0.01mg)于100ml容量瓶中,用甲苯配制成质量浓度为0.2mg/ml的标准储备液,常温密封保存,备用;
[0110]
二十八碳脂肪烷醇或三十碳脂肪烷醇标准工作溶液的配制:吸取0.5、1.0、2.5、5.0、10.0ml二十八碳脂肪烷醇或三十碳脂肪烷醇标准储备液于10ml容量瓶中,用甲苯定容,配制成0.01、0.02、0.05、0.1、0.2mg/ml的标准工作溶液,常温密封保存;
[0111]
混合脂肪烷醇标准工作溶液的配制:分别吸取0.5、1.0、2.5、5.0、10.0ml二十八碳脂肪烷醇和三十碳脂肪烷醇标准储备液于10ml容量瓶中,用甲苯定容,配制成0.01、0.02、0.05、0.1、0.2mg/ml的混合标准工作溶液,常温密封保存;
[0112]
邻苯二甲酸二癸酯工作液:准确称取50mg邻苯二甲酸二癸酯标准溶液,用甲苯配制成5mg/ml的内标工作液,冷藏保存;
[0113]
外标法:分别吸取1ml脂肪烷醇系列标准工作液,测定目标组分的色谱峰面积,以目标组分峰面积(y)为纵坐标,以其对应的质量浓度(x,mg/ml)为横坐标,作线性回归分析,如表1所示,标准曲线如图1所示;
[0114]
内标法:分别吸取1ml二十八碳脂肪烷醇或三十碳脂肪烷醇标准工作液,加入40μl邻苯二甲酸二癸酯内标工作液,混匀,测定目标峰和内标峰的色谱峰面积,以目标组分跟内标物的峰面积比(y)为纵坐标,以系列浓度(x,mg/ml)为横坐标,作线性回归分析,如表1所示,标准曲线如图2所示。
[0115]
样品测定:
[0116]
将制备实施例1的试样溶液注入气相色谱仪中,以保留时间定性,同时记录目标组分和内标物色谱峰面积的比值,根据标准曲线得到目标组分的浓度。
[0117]
结果计算:
[0118]
试样中二十八碳脂肪烷醇或三十碳脂肪烷醇含量按式(1)计算:
[0119][0120]
式(1)中:
[0121]
x——试样中二十八碳脂肪烷醇或三十碳脂肪烷醇的质量百分比含量,%;
[0122]
c——从标准曲线中得到的被测组分的浓度,单位为毫克每毫升(mg/ml);
[0123]
v——试样溶液定容体积,单位为毫升(ml);
[0124]
m——试样的质量,单位为克(g)。
[0125]
测定结果取两次测定的算术平均值,结果保留3位有效数字。
[0126]
试样中高级脂肪烷醇(二十八碳脂肪烷醇和三十碳脂肪烷醇)的含量按式(2)计算:
[0127]
x=∑xi
ꢀꢀ
(2)
[0128]
式(2)中:
[0129]
x——试样中高级脂肪烷醇(二十八碳脂肪烷醇和三十碳脂肪烷醇)的质量百分比含量,%;
[0130]
xi——试样中二十八碳脂肪烷醇或三十碳脂肪烷醇的质量百分比含量,%;
[0131]
结果保留3位有效数字。
[0132]
经测定,本发明中二十八碳脂肪烷醇和三十碳脂肪烷醇的检出限分别为0.07%和0.08%,定量限分别为0.20%和0.25%。
[0133]
(2)实施例2~11中有机溶剂提取二十八碳脂肪烷醇和三十碳脂肪烷醇的响应情况如图3所示。
[0134]
从实例2~11中发现,手动混匀后,溶剂是二氯甲烷、三氯甲烷和甲苯的样品几乎溶解;常温500w超声30min后,溶剂是二氯甲烷、三氯甲烷和甲苯的样品完全溶解,其他溶剂中,溶剂是环己烷,正己烷,沸点为60~90℃的石油醚的样品比溶剂是无水乙醇、异丙醇、甲醇和丙酮的样品溶解效果好,但也未完全溶解;在40℃下500w超声提取30min后,溶剂是无水乙醇、甲醇和丙酮的样品仍然没有完全溶解,需要更高温度超声更久才能完全溶解;在低温(2~8℃)下静置一段时间,除了溶剂为三氯甲烷的样液没有出现析出外,其他样液都出现不同程度的析出,甲苯也有少量析出,但静置至室温后,溶剂是二氯甲烷、甲苯、环己烷、沸点为60~90℃的石油醚的样液都出现复溶,其中甲苯复溶最快。另外发现,甲苯、环己烷、正己烷和沸点为60~90℃的石油醚4种溶剂中的二十八碳脂肪烷醇和三十碳脂肪烷醇的响应值很高。从溶解度看,三氯甲烷作为溶剂效果最佳,甲苯次之;从响应值看,甲苯中的响应值比三氯甲烷高很多。根据《中华人民共和国药典2005版》中溶剂毒性分类,三氯甲烷和甲苯都属于二类溶剂,但从每日允许接触量看,毒性:三氯甲烷>甲苯。从毒理学资料分析,三氯甲烷属中等毒性,具有强麻醉性,而且在人体内不能被肝脏代谢,毒性最大;甲苯属低毒类,同时甲苯很不活泼,在人体温度下不与人体蛋白质、酶、氨基酸等活性物质反应,大部分可在体内被代谢。因此,综合溶解度、响应值和毒性三方面因素,最终确定提取溶剂为甲苯。
[0135]
(3)测定实施例12中米糠脂肪烷醇样品的提取率,用来表征二十八碳脂肪烷醇或三十碳脂肪烷醇溶解情况及其响应值,各试验重复3次。提取率为提取的米糠脂肪烷醇样品中二十八碳脂肪烷醇或三十碳脂肪烷醇的峰面积占完全溶解后样液中二十八碳脂肪烷醇或三十碳脂肪烷醇峰面积的百分比。不同超声时间下二十八碳脂肪烷醇的提取率如图4所示;不同超声时间下三十碳脂肪烷醇的提取率如图5所示;不同超声温度下二十八碳脂肪烷醇的提取率如图6所示;不同超声温度下三十碳脂肪烷醇的提取率如图7所示;不同超声功率下二十八碳脂肪烷醇的提取率如图8所示;不同超声功率下三十碳脂肪烷醇的提取率如图9所示。
[0136]
由图4和图5可知,在超声温度20℃,超声功率300w的条件下,按照超声时间5,10,15,20,30min进行超声提取,随着超声时间的延长,二十八碳脂肪烷醇和三十碳脂肪烷醇的提取率都呈逐渐上升的趋势,但当时间超过20min,样品能完全溶解。
[0137]
由图6和图7可知,在超声功率300w,超声5min的条件下,按照超声温度20,25,30,40,50℃进行超声提取。随着超声温度的升高,二十八碳脂肪烷醇和三十碳脂肪烷醇的提取率都呈逐渐上升的趋势,但当温度超过40℃,样品能完全溶解。
[0138]
由图8和图9可知,在超声温度20℃,超声5min的条件下,按照超声功率300,350,400,450,500w进行超声提取。随着超声功率的升高,二十八碳脂肪烷醇和三十碳脂肪烷醇的提取率都呈逐渐上升的趋势,功率越大,溶解效果越好。
[0139]
综上可知,称样量为0.02g的样品中二十八碳脂肪烷醇和三十碳脂肪烷醇的含量高低对溶解效果有轻微影响,含量高的样品溶解速度稍微慢点,但不是影响样品溶解速度的重要因素,同时对于三十碳脂肪烷醇含量高的样品,低温时更容易析出,但恢复至常温25℃左右会复溶。
[0140]
(4)正交试验结果与分析
[0141]
选择以含量为90%的二十八碳脂肪烷醇的米糠脂肪烷醇样品中二十八碳脂肪烷醇的提取率为考察指标,设计3因素3水平正交试验,其因素水平见表2,结果见表3。
[0142]
表2正交实验设计因素及水平
[0143][0144]
表3正交试验结果
[0145][0146]
由表3可知,通过对正交试验结果进行分析,由极差r可以看出,b的极差最大,其次为a,c。极差越大,反应该因素水平变动时,米糠脂肪烷醇样品的提取率变化越大。各因素强弱顺序依次为b、a、c,即超声温度>超声时间>超声功率。从提取率中可以看出,试验2,试验3,试验5,试验8,试验9的提取率都达到了100%,考虑超声溶解后需要静置至常温再定容样液,在能达到同样效果下,选择温度低的更合适,另外从经济高效方面考虑,在能达到同样效果下,时间越短越好,因此,最终得到的最佳组合为a1b2c3,即米糠脂肪烷醇样品的提取条件为,500w,30℃水浴超声提取5分钟。在此条件下进行验证试验,样品完全溶解,得到的三种数据为:101.62%,100.74%,101.58%,平均值为101.31%。
[0147]
(5)内标筛选的结果与分析
[0148]
根据实施例1的气相色谱条件,分别将浓度为0.2mg/ml的苯甲酸苯酯和邻苯二甲酸二癸酯内标溶液进到气相色谱仪中进行分析,比较两种化合物的色谱峰。苯甲酸苯酯的色谱图见图10,邻苯二甲酸二癸酯的色谱图见图11。将40μl邻苯二甲酸二癸酯内标溶液加入到1ml样品容液中,使其浓度为0.2mg/ml,利用实施例1的气相色谱条件进行分析,其色谱图见图12。
[0149]
由图10发现苯甲酸苯酯的保留时间为3.944min,和二十八碳脂肪烷醇、三十碳脂肪烷醇的保留时间相差太大,峰面积也太小,不适宜用作内标物。
[0150]
由图11可以看出,邻苯二甲酸二癸酯的出峰介于二十八碳脂肪烷醇、三十碳脂肪烷醇之间,且能达到基线分离;同时,考虑到邻苯二甲酸二癸酯标准物质容易获得以及该组分在实际米糠脂肪烷醇产品中没有残留,选用该组分作为内标较为合适。
[0151]
(6)将标准混合储备液逐步稀释浓度,以工作站中计算的3倍和10倍信噪比(s/n)为检出限(lod)和定量限(loq)。
[0152]
根据进样浓度计算出二十八碳脂肪烷醇和三十碳脂肪烷醇的检出限分别为0.07%和0.08%,定量限分别为0.2%和0.25%。
[0153]
(7)回收率与重复性
[0154]
向样品中分别加入基本等量、2倍等量和10倍等量3个水平的二十八碳脂肪烷醇和三十碳脂肪烷醇标准溶液,按所建立的方法进行测定。每份样品进行6次平行测定,考察方法的回收率和精密度,回收率与精密度见表4。以内标法中加标量为0.60%的二十八碳脂肪烷醇和三十碳脂肪烷醇的保留时间和峰面积考察其重复性,二十八碳脂肪烷醇和三十碳脂肪烷醇的保留时间和峰面积的rsd见表5。
[0155]
表4二十八碳脂肪烷醇和三十碳脂肪烷醇的平均加标回收率和精密度(n=6)
[0156][0157][0158]
表5二十八碳脂肪烷醇和三十碳脂肪烷醇的保留时间和峰面积rsd(内标法)
[0159][0160]
由表5可知,二十八碳脂肪烷醇保留时间rsd为0.0120%,三十碳脂肪烷醇保留时间rsd为0.0204%,均小于1%;二十八碳脂肪烷醇峰面积rsd为3.17%,三十碳脂肪烷醇峰面积rsd为3.12%,均小于5%,满足重复性的要求。另外由表4可知,外标法中二十八碳脂肪烷醇和三十碳脂肪烷醇的平均回收率为95.8%~104.0%,相对标准偏差(rsd)为1.27%~3.24%;内标法中二十八碳脂肪烷醇和三十碳脂肪烷醇的平均回收率为99.3%~104.0%,相对标准偏差(rsd)为0.769%~1.67%。说明两种方式都可用于米糠脂肪烷醇中高级脂肪烷醇的测定,较外标法而言,采用内标法测定的平均加标回收率更贴近100%,且维持在
99.3%~104%之间,因此该加标实验方法可行,并且证实使用内标法测定米糠脂肪烷醇中高级脂肪烷醇含量的方法更加准确。
[0161]
(8)重现性
[0162]
选取三份米糠脂肪烷醇样品,分别送往五家检测机构,利用建立的气相色谱外标法和内标法对二十八碳脂肪烷醇和三十碳脂肪烷醇进行测定,结果见表6。
[0163]
表6五家检测机构的比对结果
[0164][0165]
重现性以相对标准偏差(rsd%)表示,rsd%越小,表明重现性越好,由表6可知,外标法测定结果的rsd在1.28%~6.45%之间,内标法测定结果的rsd在0.50%~2.96%之间,表明该实验方法具有较高的可重复性与准确性,可高效快捷的检测米糠脂肪烷醇中高级脂肪烷醇,为市场监管部门对米糠脂肪烷醇这种新食品原料的监管提供有力的技术支持。同时,采用内标法测定结果的rsd值更小,这也说明与外标法相比,内标法能够更好地校准和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。
[0166]
(9)色谱柱的选择
[0167]
色谱柱的选择对样品的分离十分重要,通常不同类型的填料对同一样品有不同的保留效果。将仪器条件设定为:进样量2μl,进样口温度300℃,检测器fid温度320℃,流速为3ml/min,进样模式为分流,分流比5:1,分流流量15ml/min,升温程序为起始温度200℃,保持1min,然后以15℃/min的速率升到260℃,保持20min。二十八碳脂肪烷醇和三十碳脂肪烷醇在中等极性db-1701(30m
×
0.320mm
×
0.25μm)色谱柱中的色谱图见图13,在弱极性hp-5(30m
×
0.320mm
×
0.25μm)色谱柱中的色谱图见图14,两个色谱图中二十八碳脂肪烷醇和三十碳脂肪烷醇的峰面积、峰高以及峰宽见表7。
[0168]
表7 db-1701和hp-5中二十八碳脂肪烷醇和三十碳脂肪烷醇的峰面积、峰高以及峰宽
[0169][0170]
由图13和14可发现,db-1701和hp-5中二十八碳脂肪烷醇和三十碳脂肪烷醇都能得到很好的分离,再通过表7比较两色谱图中二十八碳脂肪烷醇和三十碳脂肪烷醇的峰高,峰宽以及响应值发现,db-1701色谱柱中二十八碳脂肪烷醇和三十碳脂肪烷醇的峰高和响应值都小于hp-5色谱柱中二十八碳脂肪烷醇和三十碳脂肪烷醇的峰高和响应值,而峰宽又比hp-5中的色谱峰大,因此,根据二十八碳脂肪烷醇和三十碳脂肪烷醇的弱极性,以及柱子对高温的耐受性,最终选择hp-5色谱柱作为分析柱。
[0171]
(10)仪器条件
[0172]
二十八碳脂肪烷醇和三十碳脂肪烷醇属于同系物,在气相色谱分析中,对于组分沸点范围窄、化学性质类似的样品,一般选用一价升温。根据hp-5色谱柱筛选时设定的仪器参数条件,进一步完善参数设定,参数设定见表8。当仪器参数条件为:进样量2μl,进样口温度300℃,检测器fid温度320℃,流速为3ml/min,进样模式为分流,分流比5:1,分流流量15ml/min,升温程序为起始温度200℃,保持1min,然后以10℃/min的速率升到300℃,保持20min时,二十八碳脂肪烷醇和三十碳脂肪烷醇的色谱图见图15;当仪器参数条件为:进样量2μl,进样口温度300℃,检测器fid温度320℃,流速为3ml/min,进样模式为不分流,升温程序为起始温度200℃,保持1min,然后以10℃/min的速率升到300℃,保持20min时,二十八碳脂肪烷醇和三十碳脂肪烷醇的色谱图见图16;当仪器参数条件为:进样量2μl,进样口温度300℃,检测器fid温度320℃,流速为1ml/min,进样模式为不分流,升温程序为起始温度200℃,保持1min,然后以10℃/min的速率升到300℃,保持20min时,二十八碳脂肪烷醇和三十碳脂肪烷醇的色谱图见图17。
[0173]
表8仪器参数条件
[0174][0175]
通过图15至图17,可发现通过调整升温程序,进样模式以及流速后,二十八碳脂肪烷醇与三十碳脂肪烷醇的出峰时间为13.366min和15.640min,峰高为429.5和354.7,峰宽为0.0578min和0.0808min,峰面积为1575.44751和1909.22131,达到最佳响应。因此最终仪器条件为:进样量2μl,进样口温度300℃,检测器fid温度320℃,流速为1ml/min,进样模式为不分流,升温程序为起始温度200℃,保持1min,然后以10℃/min的速率升到300℃,保持20min。
[0176]
本发明通过校准曲线、线性相关系数、检出限、定量限、加标回收率,重复性以及重现性,对米糠脂肪烷醇中二十八碳脂肪烷醇和三十碳脂肪烷醇的气相色谱外标法和内标法进行方法学验证,表明两种方法都具有较好的重复性和准确度,可高效快捷的检测米糠脂肪烷醇中高级脂肪烷醇,为国家风险监测,风险评估提供技术支持,为国家制定米糠脂肪烷醇的检测方法奠定基础。同时通过两种方法的方法学验证发现,内标法比外标法的重复性和重现性好,准确度更高。
[0177]
尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例而不是全部实施例,人们还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。