一种负载20(s)-原人参二醇的固体分散体及其制备方法和应用
技术领域
1.本发明涉及一种可在胃肠道形成纳米混悬液的ppd固体分散体及其制备方法,还涉及由该ppd固体分散体制成的口服固体制剂。本发明属于医药技术领域。
背景技术:
2.20(s)-原人参二醇(protopanaxadiol,ppd)为二醇型人参皂苷元,天然来源于人参、西洋参、三七等五加科植物,以及绞股蓝等葫芦科植物。商品ppd常通过微生物酵解二醇组人参皂苷获得。ppd具有包括免疫调节、抗肿瘤、提高学习记忆、抗衰老、抗炎、抗痴呆、抗焦虑、抗抑郁、抗疲劳和促进造血等在内的广泛的药理活性。但ppd水溶性较差,在水中的平衡溶解度仅为35.24mg/l,油水分配系数(p)为46.21(logp=1.66),肠灌流模型和细胞模型表明ppd的膜渗透性好,因此ppd属于生物药剂学分类系统中属于溶解性差,渗透性好的第二类药物。
3.对于此类药物,溶出是药物吸收的限速步骤,影响药物口服生物利用度,改善药物溶出为常用的提高此类药物生物利用度的制剂方法。文献报道了采用纳米晶、自微乳和聚合物胶束等手段有效提高ppd溶出和口服生物利用度的研究(kim et al.,development of 20(s)-protopanaxadiol-loaded snedds preconcentrate using comprehensive phase diagram for the enhanced dissolution and oral bioavailability.pharmaceutics,2020,12(4):362;xia et al.,a novel drug-phospholipid complex enriched with micelles:preparation and evaluation in vitro and in vivo.int jnanomedicine,2013,8:545;chen et al.,formulation of 20(s)-protopanaxadiol nanocrystals to improve oral bioavailability and brain delivery.int j pharm,2016,497(1-2):239-47)。其中基于纳米晶技术的方法展示出较好的应用前景,但基于纳米晶技术的制剂制备复杂,制备成口服固体制剂时存在分散稳定性较差的问题。研究开发易于制造,并可保持纳米晶技术提高ppd溶出速度和口服生物利用度等优势的制剂技术存在挑战。
技术实现要素:
4.本发明的目的之一是提供一种负载20(s)-原人参二醇的固体分散体及其制备方法;
5.本发明的目的之二是提供一种基于所述的固体分散体的ppd口服固体制剂,这种制剂可在胃肠道转化为纳米混悬剂,从而提高ppd口服固体制剂的溶出速度和口服生物利用度。
6.为了达到上述目的,本发明采用了以下技术手段:
7.本发明首先是将药物、聚合物和表面活性剂溶解在乙醇溶剂中,然后采用减压干燥方法制备固体分散体。传统上,固体分散体可提高药物溶解度和溶出速度,形成过饱和药物溶液,从而提高药物在胃肠道的吸收速度和生物利用度。但药物过饱和溶液通常不稳定,
容易重结晶,因此需要选择适当的辅料延缓药物重结晶的速度,提高药物的吸收。在本发明中,我们发现ppd和聚合物辅料形成的固体分散体复溶后迅速重结晶,并产生药物沉淀。在固体分散体配方中加入表面活性剂时,药物依然可迅速重结晶,但我们意外发现当加入适量的表面活性剂为泊洛沙姆188(f68)、tpgs或sls时,可控制重结晶形成的药物粒子的粒径大小,形成平均粒径小于300nm的纳米混悬剂。进一步研究发现当表面活性剂为tpgs或sls时,纳米混悬剂在人工胃、肠液中可保持稳定至少8小时,有利于药物在胃肠道的吸收。
8.当固体分散体粉体和适当口服药用辅料混合后,可直接压制成片或填装成胶囊。所得的制剂在体外溶出试验中可迅速崩解,在15分钟内导致90%以上的药物溶出,而参比的物理混合物制剂仅溶出约11%。尤其是,固体分散体所得制剂溶出的药物浓度至少可维持8小时,保证药物在胃肠道的吸收。进一步的药代动力学研究结果发现,与可参比的物理混合物的ppd制剂相比,基于固体分散体的片剂的最大血药浓度和口服生物利用度分别是前者的6.59倍和2.54倍。
9.在上述的研究基础上,本发明提出了一种负载20(s)-原人参二醇(ppd)的固体分散体,所述的固体分散体中包含活性成分20(s)-原人参二醇以及辅料,所述的辅料由聚合物和表面活性剂组成,20(s)-原人参二醇与辅料的重量比为1:1.5~5。
10.其中,优选的,所述的固体分散体中,20(s)-原人参二醇的含量为10-40%w/w,更优选含量为15-30%w/w。
11.其中,优选的,所述的聚合物为醇溶性的聚合物,包括:聚乙烯吡咯烷酮(pvp)、乙烯基吡咯烷酮/醋酸乙烯共聚物(pvp-va)、聚乙二醇中的一种或多种混合;所述的固体分散体中,聚合物的含量为50-70%w/w;
12.所述的表面活性剂包括泊洛沙姆、维生素e聚乙二醇琥珀酸酯(tpgs)、十二烷基硫酸钠(sls)中的一种或多种混合;所述的固体分散体中,表面活性剂的含量为1-20%w/w。
13.其中,优选的,固体分散体的制备方法为:将活性成分20(s)-原人参二醇和辅料按比例溶于乙醇中,采用减压干燥法制成干燥分散体,然后将干燥分散体置于-20℃放置冷却,在干燥环境下粉碎,过筛,即得固体分散体粉体。
14.其中,优选的,将所述的固体分散体加水复溶后,可形成纳米混悬剂;纳米混悬剂的平均粒径小于500nm,优选粒径小于300nm。
15.进一步的,本发明还提出了一种含有20(s)-原人参二醇的口服固体制剂,其含有以上任一项所述的固体分散体。
16.其中,优选的,所述的口服固体制剂为片剂或胶囊,其制备方法为:通过混合固体分散体粉体与常用促崩解的辅料混合后直接压片或填装胶囊,固体分散体在片剂或胶囊中的含量为50-80%w/w。
17.其中,优选的,每粒胶囊或每片制剂中含有50-120mg的20(s)-原人参二醇。
18.其中,优选的,所述的口服固体制剂为片剂,其中含有ppd固体分散体pvp-va-tpgs-680%w/w、填充剂乳糖4%w/w、崩解剂ppvp15%w/w、润滑剂硬脂酸镁1%w/w,片剂中ppd的含量为25%w/w。
19.相较于现有技术,本发明的有益效果是:
20.1、本发明所制得的固体制剂在体外溶出试验中可迅速崩解,在15分钟内导致90%以上的药物溶出,而参比的物理混合物的制剂仅溶出约11%。尤其是,固体分散体所得制剂
溶出的药物浓度至少可维持8小时,保证药物在胃肠道的吸收。进一步的药代动力学研究结果发现,与可参比的物理混合物的ppd制剂相比,基于固体分散体的片剂的最大血药浓度和口服生物利用度分别是前者的6.59倍和2.54倍。
21.2、本发明的ppd固体分散体的制备工艺简单,易于生产,其可在胃肠道转化为稳定的纳米混悬剂,从而提高药物的溶出速度和口服生物利用度。
附图说明
22.图1为ppd固体分散体和物理混合物的体外溶出曲线
23.图2为ppd固体分散体和物理混合物以30mg/kg剂量经大鼠口服给药后的药时曲线
具体实施方式
24.下面结合具体实施例对本发明作进一步说明,但本发明不限于以下实施例。本领域技术人员应该理解的是,在不偏离本发明的精神及范围下可对本发明技术方案的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
25.实施例1:负载20(s)-原人参二醇(ppd)的固体分散体对比粉体的制备
26.称取100mg ppd,300mg聚乙烯吡咯烷酮(pvp)或乙烯基吡咯烷酮/醋酸乙烯共聚物(pvp-va)溶解在50ml无水乙醇溶剂中,旋转蒸发除去溶剂,真空干燥后将圆底烧瓶密封置于-20℃冰箱中固化20min,取出后过80目筛,置干燥器中避光保存。然后称取40mg过筛粉体至烧杯中,加入20ml人工胃液中复溶,可观测到药物迅速沉淀。
27.实施例2:负载20(s)-原人参二醇(ppd)的固体分散体的制备
28.称取100mg ppd,200mg聚乙烯吡咯烷酮(pvp)或乙烯基吡咯烷酮/醋酸乙烯共聚物(pvp-va),100mg f68溶解在50ml无水乙醇溶剂中,旋转蒸发除去溶剂,真空干燥后将圆底烧瓶密封置于-20℃冰箱中固化20min,取出后过80目筛,置干燥器中避光保存。然后称取40mg过筛粉体至烧杯中,加入20ml人工胃液中复溶,可观测到具有丁达尔效应的纳米混悬剂,通过马尔文纳米粒度仪测定后发现,纳米混悬剂的粒径分别约为200nm左右,但随着放置时间延长,药物开始沉淀。
29.实施例3:负载20(s)-原人参二醇(ppd)的固体分散体的制备
30.称取100mg ppd,200mg聚乙烯吡咯烷酮(pvp)或乙烯基吡咯烷酮/醋酸乙烯共聚物(pvp-va),100mg tpgs溶解在50ml无水乙醇溶剂中,旋转蒸发除去溶剂,真空干燥后将圆底烧瓶密封置于-20℃冰箱中固化20min,取出后过80目筛,置干燥器中避光保存。然后称取40mg过筛粉体至烧杯中,加入20ml人工胃液中复溶,可观测到具有丁达尔效应的纳米混悬剂,通过马尔文纳米粒度仪测定后发现,含pvp的纳米混悬剂的粒径约为200nm左右,而含pvp-va的粒径约为120nm。随着放置时间延长,含pvp的制剂粒径逐步增大到约400nm,而含pvp-va的粒径基本保持不变。
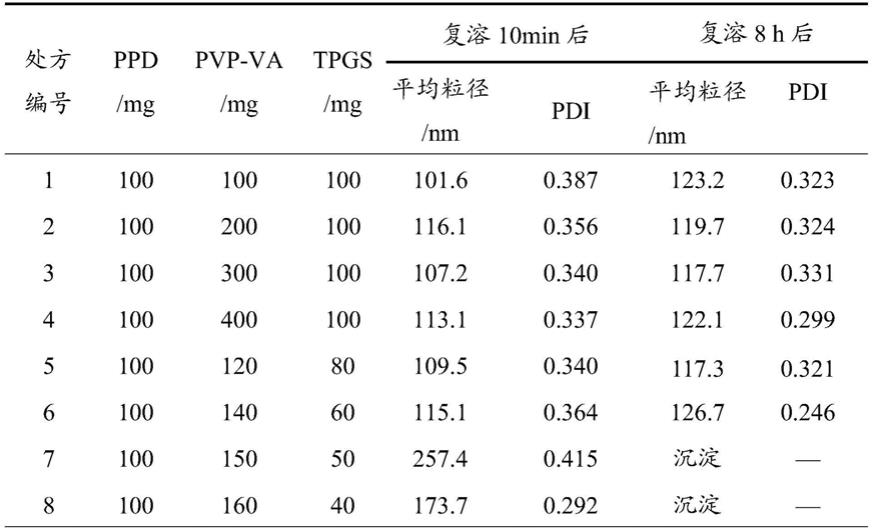
31.实施例4:tpgs浓度对纳米混悬剂的粒径和稳定的影响
32.按下表1处方组成称取ppd,乙烯基吡咯烷酮/醋酸乙烯共聚物(pvp-va)和tpgs溶解在50ml无水乙醇溶剂中,旋转蒸发除去溶剂,真空干燥后将圆底烧瓶密封置于-20℃冰箱中固化20min,取出后过80目筛,置干燥器中避光保存。然后称取400mg过筛粉体至烧杯中,
加入50ml人工胃液中复溶,发现纳米混悬剂的粒径和稳定存在tpgs浓度依赖性,结果见下表1。
33.表1
[0034][0035]
实施例5:sls浓度对纳米混悬剂的粒径和稳定的影响
[0036]
按下表2处方组成称取ppd,乙烯基吡咯烷酮/醋酸乙烯共聚物(pvp-va)和sls溶解在50ml无水乙醇溶剂中,旋转蒸发除去溶剂,真空干燥后将圆底烧瓶密封置于-20℃冰箱中固化20min,取出后过80目筛,置干燥器中避光保存。然后称取400mg过筛粉体至烧杯中,加入50ml人工胃液中复溶,发现纳米混悬剂的粒径和稳定存在sls浓度依赖性,结果见下表2。
[0037]
表2
[0038][0039]
实施例6:压片效果和崩解观察
[0040]
采用组方为ppd:pvp-va:tpgs(100:140:60)的ppd固体分散体进行预实验,以压片物料的流动性、片剂的可压性、崩解时间及外观光洁度作为评价制剂质量的指标,对分散片
的崩解剂、填充剂及润滑剂的种类及用量进行筛选。在单因素考察实验中,按基本处方进行实验:ppd固体分散体(含ppd 100mg)80%w/w、填充剂10%w/w,崩解剂9%w/w、润滑剂1%w/w。以乳糖、可压性淀粉和微晶纤维素(mcc)为填充剂,硬酯酸镁为润滑剂,崩解剂选择羧甲基淀粉钠(cms-na)、交联聚维酮(ppvp)或交联羧甲纤维素钠(cc-na)。采用直接压片法制备固体分散体片剂,每片含ppd100 mg。具体是将ppd固体分散体过80目筛,其它辅料过100目筛,按处方量分别称取主药ppd固体分散体及各种辅料,混合均匀,最后加上润滑剂过100目筛,混合均匀,检验合格后压片即可。取ppd固体分散片,照崩解时限检查法(中国药典2015年版二部附录xa),测定片剂崩解时间。预实验结果见下表3。
[0041]
表3
[0042][0043][0044]
根据预实验结果,优化的片剂处方为:ppd固体分散体pvp-va-tpgs-680%w/w、填充剂乳糖4%w/w、崩解剂ppvp15%w/w、润滑剂硬脂酸镁1%w/w,片剂中ppd的含量为25%w/w。
[0045]
实施例7:体外溶出实验
[0046]
优化后的固体分散体片剂和原药经相同处方物理混合压片的片剂进行体外溶出对比。分别取两种自制片各6片,以纯水为溶出介质,参照2015年版《中国药典》片剂溶出度测定法(浆法)进行考察。溶出介质为500ml,转速为120r
·
min-1
,温度设置为(37
±
2)℃,分别在第15、30、60、90、120、240、360、480min取样,用微孔滤膜过滤,滤过液经90%的甲醇溶液稀释后,用hplc进样分析。
[0047]
体外溶出实验结果见图1。在物理混合的片剂中,ppd原药的平均粒径约为17.4μm。从图1中的溶出结果可知,固体分散片在15min的溶出量超过了90%,而物理混合的片剂15
分钟时只有有约11%的药物溶出。不过需要注意的是,溶出的药物是以纳米混悬的形式存在,而不是药物分子形式溶出,因为溶出液采用0.22μm滤膜过滤,分散体复溶形成的纳米粒可经滤膜过滤。物理混合物的溶出药液浓度随着时间延长略有下降,而固体分散体片剂的溶出药物浓度则在8小时内保持稳定。
[0048]
实施例8:动物实验
[0049]
全部动物实验在中国医学科学院药用植物研究所动物伦理委员会指导下进行实验研究。雄性wistar大鼠(180-220g,8周),购自北京维通利华实验动物技术有限公司(scxk京2012-0001),实验前在spf动物房适应性饲养7天。所有动物均保持在控制温度(22
±
2℃),12h光照/黑暗周期,并自由饮食和饮水。
[0050]
将大鼠腹腔注射40mg
·
kg-1
戊巴比妥钠生理盐水溶液,深度麻醉后,在右嘴角向锁骨处做一条垂线,在交点上方约0.5cm的部位,用剪刀将皮肤剪开一个1cm左右的切口,钝性分离颈静脉,并将其远心端结扎,在颈静脉窦远心方向约0.5cm处剪一个小切口,沿切口处向心脏方向插入约2.5cm的聚乙烯管(id 0.5mm,od 1mm,英国portex公司),并将聚乙烯管末端沿皮下在背部导出,用肝素钠生理盐水溶液封管,结扎固定,缝合开口。术后至少恢复12h以上。所有动物给药前禁食不禁水。
[0051]
雄性wistar大鼠插管手术恢复后随机分成2组,每组6只,标号后灌胃给予优化的ppd固体分散片剂和ppd物理混合片剂。给药前,片剂按3mg
·
ml-1
的ppd浓度分散在水中,给药体积为10ml
·
kg-1
灌胃给药,相当于ppd剂量为30mg
·
kg-1
。给药后分别在15、30、45、60、120、180、240、360、720min时间点从颈静脉置留管中取全血约0.12ml放于含有抗凝剂肝素钠的ep管中,12000r/min离心5分钟收集得50μl血浆,冷冻储存待用。
[0052]
取50μl血浆置于1.5ml离心管中,加入内标地高辛(250μg/ml)10μl和40μl磷酸缓冲液,涡旋震荡5min。加入二氯甲烷1.5ml,涡旋震荡10min,12000r/min离心5min,小心吸取溶液,高纯氮气吹干挥发掉有机溶剂,残渣加入100μl 80%甲醇复溶,涡旋震荡1min,12000r/min离心5min,取上清进hplc-ms/ms分析检测。
[0053]
大鼠口服ppd固体分散片剂和物理混合片剂的药时曲线见图2。由药代结果可知,ppd固体分散片剂口服给药后可快速吸收入血,达峰时间为30min,而物理混合片剂的ppd则显示出缓慢吸收的特性,没有明显的达峰时间。ppd固体分散片剂的快速溶出显著提高了其最高血药浓度至物理混合片剂的6.59倍,同时其相对生物利用度也是其物理混合片剂的2.54倍。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。