fe3o4@cgfa复合材料的制备及应用
技术领域
1.本发明涉及复合材料技术领域,尤其涉及fe3o4@cgfa复合材料的制备及应用。
背景技术:
2.随着电子信息技术在工业、商业、军事等领域的广泛应用,无线设备在人们的日常生活中发挥了重要作用。而电子和电气设备所产生的电磁污染和电磁干扰对人们的生活和工作产生了很大的影响。因此,研制性能优良的电磁吸波材料迫在眉睫。碳材料因其比表面积大、化学稳定性好、机械强度高、密度低而成为最重要的非金属材料之一。在提纯、电池、催化、脱硫,特别是微波吸收等领域得到了广泛的应用。碳材料属于介电损耗吸收材料的类型,具有很强的研究热点,如石墨烯。gn、碳纳米纤维、碳纳米纤维、cnfs、炭黑(cb)、碳纳米管、碳纳米管及其复合材料的吸波性能得到了广泛的关注和研究。
3.煤气化作为一种极具发展前景的洁净煤技术,近年来在我国取得了长足的发展。而粉煤气化技术已成为煤气化领域的主流技术之一(被认为因煤种适应性强、碳转化率高、运行成本低而更为有效)。而在煤气化过程中,一些无机成分和少量未反应的碳会形成粉煤灰或煤气化灰分(cgfa)。cgfa是煤气化过程中不可避免的副产品,通常堆积在露天,占地面积大,利用率低,并可能造成土壤、空气、水等污染。cgfa的残余碳含量达到60%,可以作为潜在的碳基材料来源。而如何以cgfa为原料制备高吸波性能的材料有待进一步研究。
技术实现要素:
4.基于背景技术存在的技术问题,本发明提出了fe3o4@cgfa复合材料的制备及应用,制备的复合材料具有良好的电磁波吸收性能,从而促进了煤气化固体废物的资源利用。
5.本发明提出的fe3o4@cgfa复合材料制备的方法步骤如下:
6.s1:cgfa的酸化
7.s2:fe3o4@cgfa的合成
8.s21:将s1中酸化后的cgfa和聚乙烯吡咯烷酮加入蒸馏水中搅拌混匀后升温至55-65℃;
9.s22:将fecl3·
6h2o和fecl2·
4h2o分别溶解在去离子水中并混匀;
10.s23:将s22中的fecl3·
6h2o和fecl2·
4h2o溶液加入到s21中的混合液中,搅拌12-18min;
11.s24:向s23的混合液中加入浓氨水,并搅拌1.8-2.2h,控制混合液的ph为 10;
12.s25:反应结束后将制备的产物磁化,并用去离子水和无水乙醇洗至中性,经干燥制得fe3o4@cgfa。
13.优选地,所述s1中cgfa的酸化的方法步骤如下:
14.s11:向盛有cgfa的容器中加入盐酸溶液,在105℃条件下搅拌反应 1.8-2.2h;
15.s12:对s11中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,并进行干燥;
16.s13:向盛有s12中干燥后的固体颗粒的容器中加入氢氟酸溶液,在105℃条件下搅拌反应1.8-2.2h;
17.s14:对s13中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,并进行干燥;
18.s15:向盛有s14中干燥后的固体颗粒的容器中加入盐酸溶液,在105℃条件下搅拌反应1.8-2.2h;
19.s16:对s11中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,并进行干燥,制得酸化后的cgfa。
20.优选地,所述s12、s14、s16中的干燥条件为温度100-110℃、时间10-14h。
21.优选地,所述s11中盐酸溶液的浓度为5mol/l,且所述气化细渣与盐酸溶液的质量体积比为1g:8-12ml。
22.优选地,所述s13中氢氟酸溶液的质量分数为40%,且所述气化细渣与氢氟酸溶液的质量体积比为1g:8-12ml。
23.优选地,所述s15中盐酸溶液的浓度为1.19g/ml,且所述气化细渣与盐酸溶液的质量体积比为1g:8-12ml。
24.优选地,所述cgfa、聚乙烯吡咯烷酮、去离子水、fecl3·
6h2o和fecl2·
4h2o 的质量比为1:0.4-0.6:80-85:0.1-0.5:0.5-1.5。
25.优选地,所述s25中干燥的条件为:在55-65℃真空条件下干燥10-14h。
26.本发明提出的上述方法制备的fe3o4@cgfa复合材料。
27.本发明提出的fe3o4@cgfa复合材料在电磁波吸收中的应用。
28.与现有技术相比,本发明的有益技术效果:
29.本发明以cgfa为原料与铁离子复合制备的fe3o4@cgfa复合材料具有优良的电磁参数和电磁波吸收性能。实施例2制备的fe3o4@cgfa复合材料在 1.5mm厚度下,最小反射损耗(rl
min
)值可达-37.4db,有效吸收带宽(rl≦10db) 是4.16ghz(13.84-18ghz),且阻抗匹配为1.00。实施例3制备的fe3o4@cgfa 产品在吸收层厚度仅为2.0mm时,最小反射损耗(rl
min
)值可达-41.4b,有效吸收带宽(rl≦10db)为4.32ghz(13.68-18ghz)。实施例2和3制备的 fe3o4@cgfa复合材料在微波波段均表现出高性能的能量吸收。在cgfa表面加载磁性颗粒后,复合材料表现出优异的微波吸收性能,因此cgfa可以替代石墨类作为一种优良的微波吸收材料,本研究为煤气化细灰的资源高值化利用提供了有效的方向。
附图说明
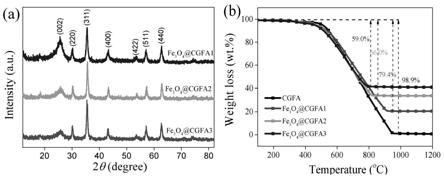
30.图1中的(a)和(b)分别为fe3o4@cgfa复合材料的xrd图和tga曲线;
31.图2中(a-c)分别为实施例1-3制备的fe3o4@cgfa复合材料的sem图像; (d)为实施例2制备的fe3o4@cgfa复合材料的edx映射图像;
32.图3为fe3o4@cgfa复合材料的透射电镜图;
33.图4为实施例2制备的fe3o4@cgfa复合材料的xps图,其中(a)为cgfa 和fe3o4@cgfa的总光谱图、(b)为fe3o4@cgfa的fe 2p图、(c)为 fe3o4@cgfa的o1s图、(d)为fe3o4@cgfa的c1s图、(e)cgfa的o1s 图、(f)cgfa的c1s图;
34.图5为本发明提出的fe3o4@cgfa复合材料的电磁参数:(a)复介电常数实部(ε
′
),
(b)复介电常数虚部(ε
″
),(c)复磁导率实部(μ
′
),(d)复磁导率虚部(μ
″
),(e)复介电常数正切值,(f)复磁导率正切值;
35.图6为本发明提出的fe3o4@cgfa复合材料的2d和3d反射损耗曲线,其中(a、b)分别为试样s1的2d和3d反射损耗曲线,(c、d)为试样s2的2d和3d 反射损耗曲线,(e、f)为试样s3的2d和3d反射损耗曲线,(g)试样s1、s2、s3 分别在厚度为1.5mm的最小反射损耗,(h)试样s1、s2、s3分别在厚度为1.5mm 的有效有效吸收带宽(rl≦10db);
36.图7为本发明提出的fe3o4@cgfa复合材料的反射损耗、λ/4模型、阻抗匹配三联图,其中(a)为试样s2的反射损耗、λ/4模型、阻抗匹配三联图,(b) 为试样s3的反射损耗、λ/4模型、阻抗匹配三联图。
具体实施方式
37.下面结合具体实施例对本发明作进一步解说。
38.主要仪器:x射线衍射仪(labx xrd-6000、日本岛津公司、日本)、激光共焦拉曼光谱仪(英国-2000,英国,英国)、x射线光电子能谱仪(escalabmk,热费希尔科学公司,美国)、场发射扫描电子显微镜(范西2000年,范有限公司,荷兰)、透射电子显微镜(jeol-2010,日本电子有限公司、日本)、矢量网络分析仪(av3629d,cetc41研究所,中国)。
39.实施例1
40.本发明提出的fe3o4@cgfa复合材料制备的方法步骤如下:
41.s1:cgfa的酸化
42.s11:向盛有50g的cgfa的容器中加入500ml盐酸溶液(5mol/l),在 105℃条件下搅拌反应1.8h;
43.s12:对s11中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,在105℃下干燥12h;
44.s13:向盛有s12中干燥后的固体颗粒的容器中加入500ml氢氟酸溶液(40% wt%),在105℃条件下搅拌反应1.8h;
45.s14:对s13中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,在105℃下干燥12h;
46.s15:向盛有s14中干燥后的固体颗粒的容器中加入500ml盐酸溶液 (1.19g/ml),在105℃条件下搅拌反应1.8h;
47.s16:对s11中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,在105℃下干燥12h,制得酸化后的cgfa。
48.s2:fe3o4@cgfa的合成
49.s21:将s1中酸化后的0.6g cgfa和0.3g聚乙烯吡咯烷酮加入50ml蒸馏水中搅拌混匀后升温至60℃;
50.s22:将0.1g的fecl3·
6h2o和0.3g的fecl2·
4h2o分别溶解在50ml去离子水中并混匀;
51.s23:将s22中的fecl3·
6h2o和fecl2·
4h2o溶液加入到s21中的混合液中,搅拌15min;
52.s24:向s23的混合液中加入浓氨水,并搅拌2h,控制混合液的ph为10;
53.s25:反应结束后将制备的产物磁化,并用去离子水和无水乙醇洗至中性,经干燥制得fe3o4@cgfa,记为s1。
54.实施例2
55.本发明提出的fe3o4@cgfa复合材料制备的方法步骤如下:
56.s1:cgfa的酸化
57.s11:向盛有50g的cgfa的容器中加入500ml盐酸溶液(5mol/l),在105℃条件下搅拌反应1.8h;
58.s12:对s11中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,在105℃下干燥12h;
59.s13:向盛有s12中干燥后的固体颗粒的容器中加入500ml氢氟酸溶液(40% wt%),在105℃条件下搅拌反应1.8h;
60.s14:对s13中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,在105℃下干燥12h;
61.s15:向盛有s14中干燥后的固体颗粒的容器中加入500ml盐酸溶液 (1.19g/ml),在105℃条件下搅拌反应1.8h;
62.s16:对s11中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,在105℃下干燥12h,制得酸化后的cgfa。
63.s2:fe3o4@cgfa的合成
64.s21:将s1中酸化后的0.6g cgfa和0.3g聚乙烯吡咯烷酮加入50ml蒸馏水中搅拌混匀后升温至60℃;
65.s22:将0.2g的fecl3·
6h2o和0.6g的fecl2·
4h2o分别溶解在50ml去离子水中并混匀;
66.s23:将s22中的fecl3·
6h2o和fecl2·
4h2o溶液加入到s21中的混合液中,搅拌15min;
67.s24:向s23的混合液中加入浓氨水,并搅拌2h,控制混合液的ph为10;
68.s25:反应结束后将制备的产物磁化,并用去离子水和无水乙醇洗至中性,经干燥制得fe3o4@cgfa,记为s2。
69.实施例3
70.本发明提出的fe3o4@cgfa复合材料制备的方法步骤如下:
71.s1:cgfa的酸化
72.s11:向盛有50g的cgfa的容器中加入500ml盐酸溶液(5mol/l),在 105℃条件下搅拌反应1.8h;
73.s12:对s11中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,在105℃下干燥12h;
74.s13:向盛有s12中干燥后的固体颗粒的容器中加入500ml氢氟酸溶液(40% wt%),在105℃条件下搅拌反应1.8h;
75.s14:对s13中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,在105℃下干燥12h;
76.s15:向盛有s14中干燥后的固体颗粒的容器中加入500ml盐酸溶液 (1.19g/ml),
在105℃条件下搅拌反应1.8h;
77.s16:对s11中的混合液进行过滤,并将残留的固体用去离子水洗涤至中性,在105℃下干燥12h,制得酸化后的cgfa。
78.s2:fe3o4@cgfa的合成
79.s21:将s1中酸化后的0.6g cgfa和0.3g聚乙烯吡咯烷酮加入50ml蒸馏水中搅拌混匀后升温至60℃;
80.s22:将0.3g的fecl3·
6h2o和0.9g的fecl2·
4h2o分别溶解在50ml去离子水中并混匀;
81.s23:将s22中的fecl3·
6h2o和fecl2·
4h2o溶液加入到s21中的混合液中,搅拌15min;
82.s24:向s23的混合液中加入浓氨水,并搅拌2h,控制混合液的ph为10;
83.s25:反应结束后将制备的产物磁化,并用去离子水和无水乙醇洗至中性,经干燥制得fe3o4@cgfa,记为s3。
84.在fe3o4@cgfa复合材料的合成过程中,fe
2
和fe
3
离子首先被cgfa的缺陷和电子-负官能团通过强静电相互作用吸引,然后加入氨水作为沉淀剂,生成铁的氢氧化物,铁的氢氧化物停留在cgfa表面。经过伴随晶粒结晶和生长的溶热反应后,自组装的fe3o4粒子被cgfa包裹,形成最终的fe3o4@cgfa复合材料。
85.参照图1,采用xrd对实施例1-3制备的复合材料的结晶结构进行分析,由图1的(a)显示,各试样衍射峰在2θ分别为30.12
°
、35.53
°
、43.18
°
、53.73
°
、 57.26
°
和62.73
°
,与fe3o4(jcpds cardno.1)的(220)、(311)、(400)、 (422)、(511)和(440)面具有较好的一致性。表明在fe3o4@cgfa复合材料中成功合成了fe3o4。cgfa在2θ=25.8
°
处(jcpds card 75-2078)出现石墨的尖锐特征峰(002),说明煤中碳在气化后转化为石墨化碳。值得注意的是,随着氧化铁负荷的增加,(002)平面的强度变弱。图1的(b)中tga曲线表明cgfa和fe3o4@cgfa分别从460℃和360℃开始失重,且cgfa、s1、s2 和s3的失重的最大温度为985℃、935℃、830℃和795℃,说明了fe3o4粒子负载在cgfa上对其具有一定的催化燃烧的效果。cgfa、s1、s2和s3在1000℃下的最大失重损失分别为98.9%、79.4%、66.0%和59.0%。通过计算1000℃下样品的总失重和剩余的cgfa重量,确定s1、s2和s3中fe3o4含量分别为19.5%、 32.9%和39.9%。
86.利用sem对实施例1-3制备的fe3o4@cgfa纳米复合材料进行了微观形貌表征。fe3o4@cgfa纳米复合材料(图2的a-c)呈不规则的片状,具有轻微聚集的fe3o4纳米颗粒均匀分布在cgfa表面,可以丰富表面积。试样s2相应的元素映射图像如图2的(d)所示。结果表明,复合材料中碳、氧、铁元素分布均匀。铁和氧的图像显示,这些元素在cgfa薄片上分布良好,其位置分布具有良好的对应关系。
87.用透射电镜研究了试样s1(a-c)、s2(d-f)和s3(g-i)的表面形貌,如图3所示,cgfa的表面覆盖着大量的fe3o4纳米颗粒,其平均尺寸约为30nm。从图3(a)和(b)、(d)和(e)、(g)和(h)可以清楚地观察到fe3o4纳米颗粒分布在形状薄的层状cgfa的表面上,但部分fe3o4纳米颗粒会产生团聚现象。值得注意的是,随着含铁量的增加,所产生的fe3o4纳米颗粒逐渐增加,结块程度也越来越严重。hrtem图像(图3的(c)、(f)和(i))清楚地显示,0.29nm和0.24nm的面间距与fe3o4的(220)面和(311)面一致。在此基础上,具有明确的盘状形貌的fe3o4@
cgfa有利于提高电磁吸波材料的电化学性能。
88.为了进一步测定cgfa和fe3o4@cgfa的元素价和化学成分,以试样s2 为例,进行了xps测定。从图4的(a)中可以看出,该总光谱图验证了cgfa 的c、o和fe3o4@cgfa的c、o和fe的存在,表明合成产物的纯度高。图4 的(b)中的fe 2p3/2能谱可拟合出三个明显的峰,分别在709.8、711.4和713.1ev 处,对应于fe
2
和fe
3
。计算得出fe
3
/fe
2
的比例为1.84。从图4的(c)可以看出,o 1s谱在530.5ev和532.0ev分为两个主峰,证实了fe3o4的形成和-oh 的存在。图4(d)显示了fe3o4@cgfa复合材料的碳1s谱。结合能为284.4ev、 285.6ev、287.0ev和290.2ev的峰分别来自于c-c/c=c、c-o、o-c=o和成环π-π*键。这说明cgfa表面存在与氧有关的官能团。表1显示了cgfa和 fe3o4@cgfa材料的重量百分比和原子百分比。根据cgfa的o/c重量比和原子比分别为0.074和0.063,而fe3o4@cgfa的o/c重量比和原子比分别增加到 0.332和0.249,这是由于fe3o4的成功负载造成的。综上所述,xps结果揭示了 fe3o4@cgfa复合材料中fe
3
、fe
2
、o和c的共存。
89.表1 cgfa和fe3o4@cgfa的重量百分比和原子百分比
[0090][0091]
一般来说,吸收剂的emw吸收行为主要取决于电磁参数(ε
′
、ε
″
、μ
′
和μ
″
)。虚部(ε
″
、μ
″
)分别表示磁损耗或介电损耗,实部(ε
′
、μ
′
)分别与存储的磁能或电能有关。图5的(a)-(d)描述了合成的fe3o4@cgfa纳米复合材料的随频率变化的电磁参数。如图5(a)所示,随着频率的增加,fe3o4@cgfa纳米复合材料的ε
′
呈逐渐下降的趋势,表现出频率色散行为,这有助于电磁能量的衰减。此外,随着含铁量的增加,ε
′
逐渐减小,这与低碳含量有关。从图5的(b) 可以看出,所合成的纳米复合材料的ε
″
具有类似的趋势,试样s1在整个频率范围内的值最大,这可以归因于其高导电性。图5(c)和(d)描述了fe3o4@cgfa 纳米复合材料的μ
′
和μ
″
与频率曲线。μ
′
和μ
″
值呈下降趋势。此外,在818ghz内μ
″
值均为负值,说明存在辐射现象。为了比较介电损耗性能,我们绘制了介电损耗正切(tanε=ε
″
/ε
′
))与频率的曲线,如图5的(e)所示。介电损耗主要来源于极化损耗和传导损耗,极化损耗包括电性、离子性、偶极性和界面极化损耗,而导电损耗主要来源于电导率损耗。特别是试样s1和试样s2的tanε与频率的曲线形状相似,而试样s2在6-10ghz时表现出不同的趋势,说明负载含量在一定程度上影响了fe3o4@cgfa纳米复合材料的介电损耗能力。为了研究磁损性能,绘制了磁损切线(tanμ=μ
″
/μ')与频率的曲线。图5的(f)中三个试样的tan μ与频率的曲线与μ
″
的变化趋势相似。此外,不同fe3o4负载量的fe3o4/cgfa 纳米复合材料的tanε明显大于tanμ。这表明,介电损耗是纳米复合材料电磁能量衰减的主要机制。
[0092]
为了考察fe3o4的添加量对制备的fe3o4@cgfa纳米复合材料emw吸收行为的影响,绘制了厚度为1.5-5.0mm的fe3o4@cgfa在2-18ghz范围内的2d 和3d rl-频率的曲线。值得注意的是,试样s3与其他两种试样相比表现出最好的emw吸收行为(图6的(e)、(f)),即在2.0mm时rl
min
为-41.4db,当厚度为1.5mm时,最大eab可达到4.32ghz。如图6的(a)、(b)所示,
试样s1表现出较差的emw吸收行为,即当厚度为1.5mm时,其在11.92ghz时的rl
min
为-9.6db。从图6的(c)、(d)可以看出试样s2的emw吸收能力较好,当厚度为1.5mm时rl
min
为-37.3db,最大的eab可以达到4.16ghz (18-13.84ghz),可以实现几乎所有ku波段的(12-18ghz)吸收,为了更直观地看到rl和eab随试样的变化,图6(g)和(h)分别给出了|rlmin|和 eab-fe3o4@cgfa曲线和直方图。随着fe3o4添加量的增加,薄厚度为1.5mm 时|rlmin|值呈现先增后减的趋势。试样s2和s3的eab均大于4。综上所述,与试样s1相比,试样s2和s3表现出了良好的emw吸收行为,说明一定量的 fe3o4负载量可以有效提高emw的吸收能力。
[0093]
为了有效地吸收电磁波,吸收或吸收结构材料应满足以下要求:材料的电磁波入射表面上完全可以进入其内部,减少表面的反射到最大程度,叫做阻抗匹配特性的材料。因此,可以根据阻抗匹配特性(z)和拟合λ/4波长匹配模型来研究阻抗匹配机理。一个理想的吸波器应该表现出零回流和1.0的目标,表示所有的波可以进入吸波器而没有回流。因此,当z的计算值接近1时,阻抗匹配程度更好,微波吸收性能更强。并将反射损耗曲线峰值对应的频率点用波长匹配模型进行匹配。图7给出了2种不同厚度复合材料在2-18ghz时的阻抗匹配特性和不同厚度的微波/4波长匹配模型。可以看出,随着厚度的增加,试样s1、s2 和s3的反射损失曲线峰值对应的频率点向低频偏移,结果与mr/4波长匹配模型一致。在厚度为1.5mm和2.0mm时,试样s2的z值分别为1.00和1.12,在厚度为1.5mm和2.0mm时,试样s3的z值分别为0.90和0.98,证明了试样s2 和s3的复合材料具有很好的阻抗匹配。
[0094]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。