1.本发明属于药物递送系统技术领域,具体涉及一种糖基聚合物及其制备方法和用途。
背景技术:
2.癌(cancer)是指起源于上皮组织的恶性肿瘤。数据显示,2020年全球新发癌症病例1929例,其中男性1006万例,女性923万例;2020年全球癌症死亡病例996万例,其中男性553万例,女性443万例。癌症发病率和死亡率高,已成为全球严重威胁人类健康的疾病之一,也是当前基础及临床研究的热点和难点之一。根据2020年最新数据显示,全球发病率前十的癌症分别是乳腺癌、肺癌、结直肠癌、前列腺癌、胃癌、肝癌、宫颈癌、食管癌、甲状腺癌、膀胱癌。目前癌症的治疗方式主要包括手术、放疗、化疗和免疫治疗等。尽管这些治疗方式的前期治疗效果较好,但仍然存在肿瘤转移、易复发、机体损伤、免疫受损等问题。
3.近年来,光动力治疗(photodynamic therapy,pdt)作为一种新兴的治疗方式。光动力治疗通过光敏剂在特定激光照射后产生的大量活性氧物质(reactive oxygen species,ros)来诱导肿瘤细胞凋亡并造成dna损伤。pdt具有高选择性、低系统性毒副作用、可重复治疗和不易产生耐受等优点,越来越受到研究者的关注。然而,大多数pdt使用的光敏剂存在水溶性差、缺乏靶向性和ros产率较低等问题,在肿瘤部位聚集不够,对肿瘤杀伤作用不强,在癌症治疗应用中受到限制。
4.如何提高光敏剂在肿瘤部位的聚集浓度,并增强pdt对肿瘤的杀伤作用,增强pdt对癌症治疗的效果,是目前癌症治疗中亟需解决的问题。
技术实现要素:
5.为了解决上述问题,本发明提供了一种糖基聚合物及其制备方法和用途。
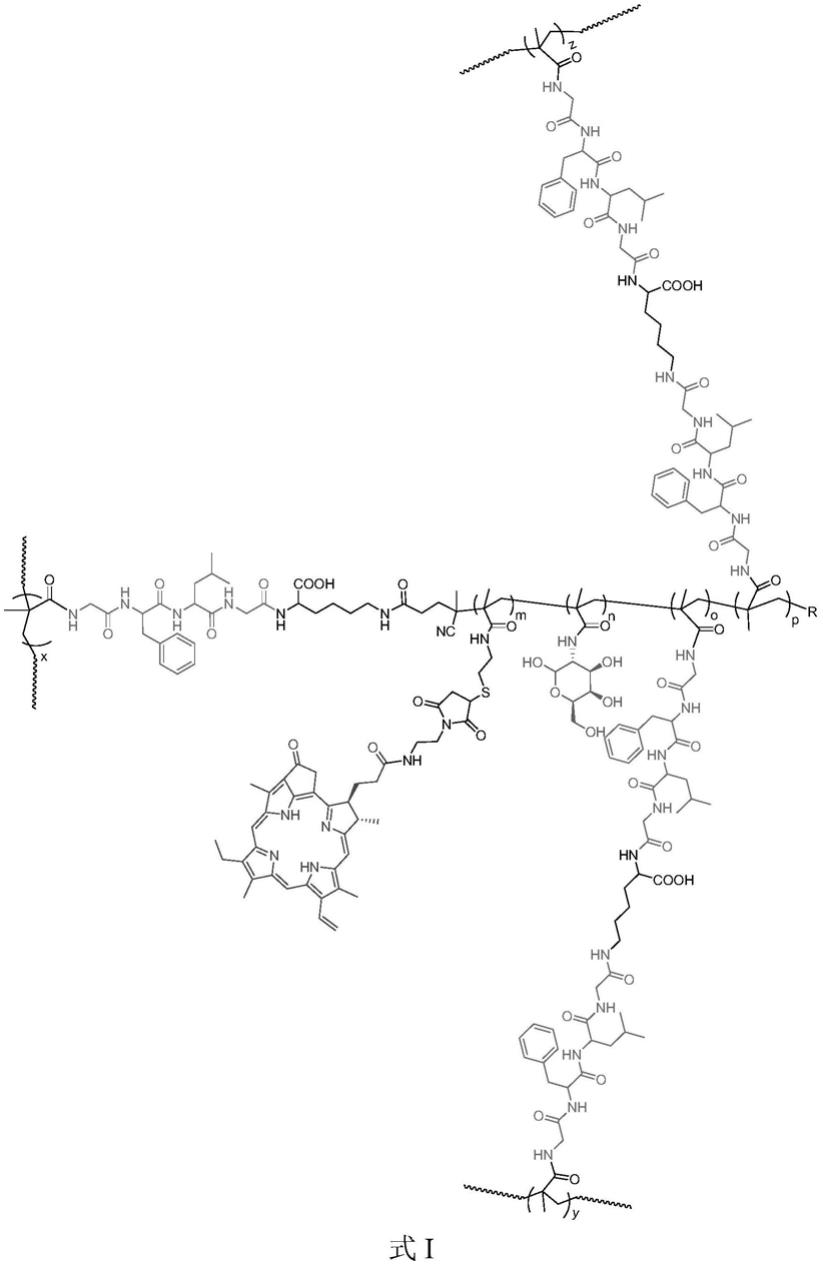
6.本发明提供了一种糖基聚合物,所述聚合物的结构为式i所示:
[0007][0008]
其中,
[0009]
(x y z):o:p:n:m的摩尔比为2.9:0.5:0.5:260:1;
[0010]
r选自
[0011]
进一步地,前述的糖基聚合物是由如下重量配比的原料制备而成:b
‑
gala
‑
sh 100~500份、maleimide
‑
ppa 1~100份;
[0012]
所述maleimide
‑
ppa的结构式为:
[0013]
所述b
‑
gala
‑
sh由如下重量配比的原料制备而成:b
‑
gala
‑
pyss 1000~1500份、二硫苏糖醇1000~1500份;
[0014]
所述b
‑
gala
‑
pyss由如下重量配比的原料制备而成:ma
‑
d
‑
galactosamine 1000~1500份、ma
‑
pyss 100~200份、ma
‑
gflgkglfg
‑
ma 10~50份、ma
‑
gflgk
‑
cta 50~100份;
[0015]
所述ma
‑
d
‑
galactosamine的结构为
[0016]
所述ma
‑
pyss的结构为
[0017]
所述ma
‑
gflgkglfg
‑
ma的结构为
[0018][0019]
所述ma
‑
gflgk
‑
cta的结构为
[0020][0021]
进一步地,所述糖基聚合物由如下重量配比的原料制备而成:b
‑
gala
‑
sh 500份、maleimide
‑
ppa 75份;
[0022]
和/或,所述b
‑
gala
‑
sh由如下重量配比的原料制备而成:b
‑
gala
‑
pyss 1300份、二硫苏糖醇1000份;
[0023]
和/或,所述b
‑
gala
‑
pyss由如下重量配比的原料制备而成:ma
‑
d
‑
galactosamine 1446份、ma
‑
pyss 100份、ma
‑
gflgkglfg
‑
ma 23.5份、ma
‑
gflgk
‑
cta 53份。
[0024]
进一步地,所述b
‑
gala
‑
sh的制备方法包括如下步骤:
[0025]
将b
‑
gala
‑
pyss与二硫苏糖醇反应得到。
[0026]
进一步地,所述b
‑
gala
‑
pyss的制备方法包括如下步骤:
[0027]
将ma
‑
d
‑
galactosamine、ma
‑
pyss、ma
‑
gflgkglfg
‑
ma和ma
‑
gflgk
‑
cta溶于溶剂中,在引发剂作用下反应,冻干,即得。
[0028]
进一步地,
[0029]
所述溶剂为水和甲醇混合溶液;
[0030]
和/或,所述反应前除氧;
[0031]
和/或,所述反应为避光反应;
[0032]
优选地,所述水和甲醇体积比为1:4;
[0033]
和/或,所述引发剂为va044;
[0034]
和/或,所述反应为40~50℃油浴反应10~12小时。
[0035]
本发明还提供了一种制备前述的糖基聚合物的方法,它包括如下步骤:
[0036]
(1)将b
‑
gala
‑
sh溶于水中,加入dmso,配制成b
‑
gala
‑
sh/dmso混合液;
[0037]
(2)将maleimide
‑
ppa溶于dmso中,加入到b
‑
gala
‑
sh/dmso混合液中反应;
[0038]
(3)将步骤(2)的反应液纯化,即得;
[0039]
优选地,
[0040]
步骤(1)中,所述水和dmso的体积比为1:(1~5);
[0041]
和/或,步骤(2)中,所述反应为室温下避光反应。
[0042]
本发明还提供了前述的糖基聚合物在制备光敏剂的药物中的用途;
[0043]
优选地,所述光敏剂是光动力治疗肿瘤的药物。
[0044]
本发明还提供了前述的糖基聚合物在制备药物载体中的用途;
[0045]
优选地,所述药物载体用于包载的药物为抗肿瘤药物;
[0046]
更优选地,所述抗肿瘤药物为奥拉帕尼。
[0047]
本发明还提供了一种药物,它是由前述的糖基聚合物为活性成分,或者前述的糖基聚合物为载体包载药物为活性成分,加上药学上可接受的辅料或辅助性成分制备而成的制剂;
[0048]
优选地,所述包载的药物为抗肿瘤药物;
[0049]
更优选地,所述包载的药物为奥拉帕尼。
[0050]
本发明提供了一种糖基聚合物bsp,该糖基聚合物克服了现有pdt光敏剂肿瘤部位聚集不够,对肿瘤杀伤作用不强的缺点,可以在肿瘤部位聚集,具有良好的靶向作用;其具有光动力效应,作为光敏剂用于光动力治疗ros产生效率高,对肿瘤细胞杀伤作用强。同时,该糖基聚合物还可以作为给药系统包载药物,特别是肿瘤药物,如奥拉帕尼,用于肿瘤治疗,效果优异。本发明糖基聚合物用于制备治疗肿瘤的药物具有优良的应用前景。
[0051]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0052]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
[0053]
图1为组织蛋白酶b敏感的功能化链转移剂ma
‑
gflgk
‑
cta的结构式及其制备路线。
[0054]
图2为组织蛋白酶b敏感的功能化交联剂ma
‑
gflgkglfg
‑
ma的结构式及其制备路线。
[0055]
图3为branched
‑
gflg
‑
poly
‑
d
‑
galactosamine
‑
s
‑
ppa(bsp)的合成路线及结构示意图。
[0056]
图4为聚合物前体branched
‑
gflg
‑
poly d
‑
galactosamine
‑
pyss的氢谱(溶剂为d6
‑
dmso)。
[0057]
图5为聚合物前体branched
‑
gflg
‑
poly d
‑
galactosamine
‑
sh的氢谱(溶剂为d6
‑
dmso)。
[0058]
图6为聚合物branched
‑
gflg
‑
poly d
‑
galactosamine
‑
s
‑
ppa的氢谱(溶剂为d6
‑
dmso)。
[0059]
图7为化合物maleimide
‑
hexyl
‑
ppa的结构式及其合成路线。
[0060]
图8为聚合物branched
‑
gflg
‑
poly d
‑
galactosamine
‑
s
‑
hexyl
‑
ppa的氢谱(溶剂为d6
‑
dmso)。
[0061]
图9为线性糖基聚合物光动力治疗体系的合成路线。
[0062]
图10为聚合物前体linear
‑
poly d
‑
galactosamine
‑
pyss的氢谱(溶剂为d2o)。
[0063]
图11为聚合物前体linear
‑
poly d
‑
galactosamine
‑
sh的氢谱(溶剂为d6
‑
dmso)。
[0064]
图12为聚合物linear
‑
poly d
‑
galactosamine
‑
s
‑
ppa的氢谱(溶剂为d6
‑
dmso)。
[0065]
图13为基于hpma的可降解支化/交联聚合物光动力治疗体系的合成路线。
[0066]
图14为聚合物前体branched
‑
gflg
‑
poly hpma
‑
pyss的氢谱(溶剂为d2o)。
[0067]
图15为聚合物前体branched
‑
gflg
‑
poly hpma
‑
sh的氢谱(溶剂为d2o)。
[0068]
图16为聚合物branched
‑
gflg
‑
poly hpma
‑
s
‑
ppa的氢谱(溶剂为d2o)。
[0069]
图17为bsp、bshp和lsp的化学结构式、自组装示意图及tem结果(标尺:200nm)。
[0070]
图18为bsp、bshp和lsp的dls检测结果:a为粒径检测结果;b为zeta电位检测结果。
[0071]
图19为bsp、bshp及lsp聚合物溶液中加入单线态氧荧光探针(sosg),随着激光照射时间的增加的荧光强度的变化情况。
[0072]
图20为bsp、bshp和lsp的体外细胞毒性实验结果。
[0073]
图21为bsp、lsp和bshp三组聚合物在肿瘤小鼠模型中分布图像及其肿瘤部位的荧光信号统计结果:a为分布图像;b为肿瘤部位的荧光信号统计结果。
[0074]
图22为通过hplc测量聚合物lsp、bsp、bshp及bhsp包载奥拉帕尼的含量,其含量通过标准曲线计算得到;hrms结果表明包载的奥拉帕尼为原药,不影响其活性。
[0075]
图23为bspo的检测结果:a为tem图像;b为dls粒径检测结果;c为zeta电位检测结果。
[0076]
图24为bsp、bspo、ppa和奥拉帕尼的荧光光谱图。
[0077]
图25为bsp、bspo、ppa和奥拉帕尼的紫外光谱图。
[0078]
图26为bsp和bspo溶液中加入单线态氧荧光探针(sosg),随着激光照射时间的增加,其荧光强度的变化情况。
具体实施方式
[0079]
本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得。
[0080]
鼠源4t1乳腺癌细胞株购自中国科学院细胞库(上海),使用含1%双抗与10%胎牛血清的rpmi 1640培养基培养,培养箱条件为5%co2,95%空气,37℃,恒湿环境。所有动物实验均严格按照医院伦理委员会批准的动物研究指南进行。实验用雌性balb/c小鼠购自成都达硕实验动物有限公司。用4t1细胞建立皮下瘤肿瘤模型,并用于体内分布和成像研究。
[0081]
实施例1、本发明糖基聚合物的制备
[0082]
化合物maleimide
‑
ppa、ma
‑
pyss和ma
‑
d
‑
galactosamine按文献1和文献2报道的方法合成。
[0083]
文献1(pan d,zheng x,zhang q,et al.dendronized
‑
polymer disturbing cells'stress protection by targeting metabolism leads to tumor vulnerability[j].advanced materials,2020,32:1907490.)报道了maleimide
‑
ppa和ma
‑
pyss的合成方法,具体合成路线如下:
[0084]
maleimide
‑
ppa:
[0085][0086]
ma
‑
pyss:
[0087][0088]
文献2(wartchow c a,wang p,bednarski m d,et al.carbohydrate protease conjugates:stabilized proteases for peptide synthesis[j].the journal of organic chemistry,1995,60:2216
‑
2226.)报道了ma
‑
d
‑
galactosamine的合成方法。
[0089]
maleimide
‑
ppa的结构为:其中ppa为光敏剂焦脱镁叶绿酸
‑
a(pyropheophorbide a)结构部分。
[0090]
ma
‑
pyss的结构为:
[0091]
ma
‑
d
‑
galactosamine的结构为:
[0092]
1、酶敏感的功能化链转移剂ma
‑
gflgk
‑
cta的制备
[0093]
酶敏感的功能化链转移剂ma
‑
gflgk
‑
cta的合成路线如图1所示。ma
‑
gflgk
‑
cta按文献3报道的类似方法合成(文献3:sun l,li x,wei x,et al.stimuli
‑
responsive biodegradable hyperbranched polymer
–
gadolinium conjugates as efficient and biocompatible nanoscale magnetic resonance imaging contrast agents[j].acs applied materials&interfaces,2016,8:10499
‑
10512.)。具体合成方法如下:
[0094]
ma
‑
gflg
‑
oh(4.6g,10mmol)、hobt(1.49g,11mmol)与缩合剂hbtu(4.26g,11mmol)置于圆底烧瓶中,氮气保护,冰浴下加入超干的dmf(50ml)使其溶解。将dipea(6.7ml,40mmol)滴加到体系中反应0.5小时。称取h
‑
lys(o
t
bu)
‑
fmoc
·
hcl(4.62g,10mmol)加入到体系中,混合溶液在冰浴下反应0.5小时后回至室温反应10小时。加入450ml乙酸乙酯(ea)溶解,再依次用饱和碳酸氢钠溶液(100ml
×
3)、1m稀盐酸(100ml
×
3)和饱和氯化钠溶液(100ml
×
3)洗涤,收集有机相,无水硫酸镁干燥后浓缩,4℃静置后析出晶状白色固体ma
‑
gflgk(o
t
bu)
‑
nhfmoc(6.51g,7.51mmol,产率75.1%)。1h nmr(400mhz,δin ppm),
13
c nmr(100mhz,d6‑
dmso,δin ppm),lc
‑
ms(es ):m/z=867.2[m h]
。maldi
‑
hrms:m/z=889.4474[m na]
。
[0095]
冰浴下,ma
‑
gflgk(o
t
bu)
‑
nhfmoc(4.33g,5mmol)用40ml dcm/tfa(v:v=1:9)的混合溶液溶解,回至室温反应。tlc监测,待反应完全后旋转蒸发除去溶剂,加入乙醚后析出白色固体粉末,得到的白色固体粉末用乙醚洗涤两次并干燥得产物ma
‑
gflgk(oh)
‑
nhfmoc
(3.61g,4.45mmol,产率89.0%)。1h nmr(400mhz,δin ppm),
13
c nmr(100mhz,d6‑
dmso,δin ppm),lc
‑
ms(es ):m/z=811.3[m h]
,834.3[m na]
,maldi
‑
hrms:m/z=833.3842[m na]
。
[0096]
以ma
‑
gflgk(oh)
‑
nhfmoc为原料,采用固相多肽合成法,经以下步骤合成功能单体ma
‑
gflgk
‑
cta。ma
‑
gflgk(oh)
‑
nhfmoc(1.62g,2mmol)与dipea(0.67ml,4mmol)溶于10ml dmf后加入氯三苯甲基氯树脂(5.0g,1.15mmol/g)反应2小时。将树脂移至聚丙烯管中,用dcm:meoh:dipea(v:v:v=17:2:1)(100ml)的混合溶液冲洗3次,再分别用dcm和dmf冲洗3次。fmoc保护基团用50ml含20%哌啶的dmf处理三次后加入氰基戊酸二硫代苯甲酸(cta
‑
cooh,2.79g,10mmol)、dic(1.26g,10mmol)和hobt(1.35g,10mmol),反应12小时。产物用tfe/dcm(v:v=3:7)在室温下处理2小时并通过过滤除去树脂。将母液减压浓缩,溶于少许甲醇后加入到乙醚中沉淀纯化,再通过高效液相色谱进一步纯化,得到粉红色固体产物ma
‑
gflgk
‑
cta(892mg,1.05mmol,产率52.5%)。1h nmr(400mhz,δin ppm),
13
c nmr(100mhz,d6‑
dmso,δin ppm),lc
‑
ms(es ):m/z=851.3[m h]
。maldi
‑
hrms:m/z=848.3408[m
‑
h]
‑
,870.3181[m na
‑
2h]
‑
。
[0097]
2、酶敏感的功能化交联剂ma
‑
gflgkglfg
‑
ma的制备
[0098]
组织蛋白酶b敏感的功能化交联剂ma
‑
gflgkglfg
‑
ma合成路线如图2所示。ma
‑
gflgkglfg
‑
ma按文献3报道的类似方法合成(文献3:sun l,li x,wei x,et al.stimuli
‑
responsive biodegradable hyperbranched polymer
–
gadolinium conjugates as efficient and biocompatible nanoscale magnetic resonance imaging contrast agents[j].acs applied materials&interfaces,2016,8:10499
‑
10512.)。具体合成方法如下:
[0099]
bocnh
‑
gflg
‑
oh(2.46g,5mmol)、hobt(743mg,5.5mmol)和hbtu(2.13g,5.5mmol)置于圆底烧瓶,并在氮气保护下加入20ml超干dmf。冰浴下加入dipea(3.35ml,20mmol)反应0.5小时。向体系中加入h
‑
lys(och3)
‑
oh
·
2hcl(583mg,2.5mmol)后回至室温反应20小时。将反应液加入到250ml乙酸乙酯中,依次用饱和碳酸氢钠溶液(20ml
×
3)、1m稀盐酸(20ml
×
3)和饱和氯化钠溶液(20ml
×
3)洗涤,收集有机相,无水硫酸镁干燥后浓缩,置于4℃结晶得到白色固体产物bocnh
‑
gflgkglfg
‑
nhboc(1.62g,1.46mmol,产率58.4%)。1h nmr(400mhz,δin ppm),
13
c nmr(100mhz,δin ppm),lc
‑
ms(es ):m/z=1109.4[m h]
,maldi
‑
hrms:m/z=1131.6045[m na]
。
[0100]
将bocnh
‑
gflgkglfg
‑
nhboc(1.33g,1.2mmol)置于氮气保护的圆底烧瓶中,冰浴下加入25ml dcm/tfa(v:v=1:1)混合溶液,回至室温反应12小时后旋去溶剂,加入无水乙醚两次后抽干得到白色的固体中间产物,将其溶于100ml h2o/acn(v:v=4:1)的混合溶液中,冰浴下滴加1m的naoh溶液调节ph至7
‑
8,然后将甲基丙烯酰氯(0.3ml,3mmol)溶于乙腈(acn)后滴加到体系中,与此同时缓慢滴加1m的naoh溶液使体系维持在ph=10,冰浴下反应1小时后置于室温反应4小时,将反应液加入200ml乙酸乙酯中,用1m的稀盐酸调节ph至2
‑
3,水相用ea萃取三次合并有机相用氯化钠溶液(20ml
×
3)洗涤,最后用无水硫酸镁干燥后浓缩,所得浓缩液置于4℃结晶得到白色固体产物ma
‑
gflgkglfg
‑
ma(700mg,0.68mmol,产率56.7%)。1h nmr(400mhz,δin ppm),
13
c nmr(100mhz,δin ppm),lc
‑
ms(es ):m/z=516.3[m 2h]
2
,1031.4[m h]
。maldi
‑
hrms:m/z=1031.5506[m h]
,1053.5377[m na]
,1029.5364[m
‑
h]
‑
。
[0101]
3、可降解的支化/交联糖基聚合物光动力治疗体系的构建与制备
[0102]
可降解的支化/交联糖基聚合物光动力治疗体系的合成路线如图3所示。具体合成方法如下:
[0103]
单体ma
‑
d
‑
galactosamine(1446mg,4.63mmol)、ma
‑
pyss(100mg,0.39mmol)、交联剂ma
‑
gflgkglfg
‑
ma(23.5mg,22.8μmol)、链转移剂ma
‑
gflgk
‑
cta(53.0mg,62.4μmol)与引发剂va044(6.7mg,20.8μmol)溶于h2o/ch3oh(7.2ml,1:4,v/v)的混合溶液中,将聚合瓶避光置于冰浴中并通氩气鼓泡除氧45分钟后将其密闭,避光置于45℃油浴反应12小时,用液氮淬灭反应后将反应液低温透析冻干,得淡红色固体branched
‑
gflg
‑
poly d
‑
galactosamine
‑
pyss(b
‑
gala
‑
pyss,1480mg,产率91.2%)。聚合物前体branched
‑
gflg
‑
poly d
‑
galactosamine
‑
pyss(b
‑
gala
‑
pyss)的氢谱如图4所示。
[0104]
将1300mg b
‑
gala
‑
pyss溶于10ml水溶液,加入1000mg二硫苏糖醇(dtt)处理过夜后透析(4℃,mwco 3.5kda)2天,样品用水相滤头处理后冻干得到白色固体粉末branched
‑
gflg
‑
poly d
‑
galactosamine
‑
sh(b
‑
gala
‑
sh,1150mg,产率88.5%)。聚合物前体branched
‑
gflg
‑
poly d
‑
galactosamine
‑
sh(b
‑
gala
‑
sh)的氢谱如图5所示。
[0105]
branched
‑
gflg
‑
poly d
‑
galactosamine
‑
s
‑
ppa(bsp)按如下方法制备:500mg b
‑
gala
‑
sh溶于5ml ro h2o后加入10ml dmso配制成b
‑
gala
‑
sh/dmso混合液,75mg maleimide
‑
ppa溶于2ml dmso后搅拌下滴加到b
‑
gala
‑
sh/dmso混合液中将得到的墨绿色溶液,于室温环境避光反应过夜后透析(避光,mwco 3.5kda)2天除去dmso,离心(7000rpm
×
5min)后取上清液经水相滤头(0.45μm)过滤,滤液冻干得黑绿色固体粗品(504mg,产率87.7%)。将粗品用2ml水溶解后滴加到200ml乙腈中出现大量沉淀,离心(10000rpm
×
5min)处理后收集固体残留物并重复处理纯化三次,得到的固体再次溶于20ml去离子水后冻干得产物(bsp,420mg)。聚合物branched
‑
gflg
‑
poly d
‑
galactosamine
‑
s
‑
ppa(bsp)的氢谱如图6所示。
[0106]
bsp的氨基酸分析结果如表1所示。
[0107]
表1.糖基支化/交联聚合物bsp的氨基酸分析结果
[0108][0109]
一般而言,聚合物载体在生物体内有效代谢的肾阈值约为50kda,因而需要保证降解后的聚合物主链分子量低于这一水平。研究发现,本发明糖基聚合物材料能在酶作用下迅速降解,4小时后的降解产物(mn=32.4
×
103,mw=46.7
×
103,pdi=1.44)有着较好的均一性,其分子量低于肾阈值。
[0110]
对照例1、branched
‑
gflg
‑
poly d
‑
galactosamine
‑
s
‑
hexyl
‑
ppa(bshp)的制备
[0111]
1、功能化光敏剂maleimide
‑
hexyl
‑
ppa的制备
[0112]
功能化光敏剂的合成路线如图7所示。
[0113]
称取马来酰亚胺丙酸(maleimide
‑
cooh,1.17g,6.9mmol)和缩合剂hatu(3.74g,9.85mmol)溶于10ml dmf,冰浴下加入diea(3.2ml,18.3mmol)反应5分钟后再将n
‑
boc
‑
1,6
‑
己二胺盐酸盐(1.80g,8.3mmol)加入体系反应2小时。将50ml饱和碳酸氢钠溶液和30ml dcm
加入到反应液中,收集有机相并将水相用dcm萃取三次,合并有机相并用饱和碳酸氢钠溶液、稀盐酸和饱和氯化钠溶液洗涤两次,无水硫酸镁干燥后去除溶剂得到黄色固体粗产物。过柱纯化(ch3oh:dcm=1:20,v/v)后用高效液相色谱进一步分离纯化,得到无色液体产物maleimide
‑
hexyl
‑
nhboc(1.66g,产率65.4%)。1h nmr(400mhz,δin ppm),
13
c nmr(100mhz,δin ppm),lc
‑
ms(es ):m/z=368.3m/z[m h]
,390.2m/z[m na]
。maldi
‑
hrms:m/z=390.2000[m na]
。
[0114]
在0℃下,将maleimide
‑
hexyl
‑
nhboc(220mg,0.60mmol)溶于10ml dcm中后加入10ml tfa,反应过夜后除去溶剂,残留物通过无水乙醚处理得到固体粉末。将上步所得的固体粉末、焦脱镁叶绿酸a(270mg,0.50mmol)与缩合剂hatu(285mg,0.75mmol)溶于dcm中并加入diea(0.23ml,1.33mmol),反应液在室温下反应1.5小时后旋去溶剂,过柱纯化(dcm:ch3oh=30:1
‑
20:1,v/v)得到黑色固体粉末maleimide
‑
hexyl
‑
ppa(237mg,产率50.5%)。1h nmr(400mhz,δin ppm),lc
‑
ms(es ):m/z=784.4m/z[m h]
。maldi
‑
hrms:m/z=806.3995[m h]
,m/z=822.3678[m k]
。
[0115]
2、branched
‑
gflg
‑
poly d
‑
galactosamine
‑
s
‑
hexyl
‑
ppa(bshp)的制备
[0116]
branched
‑
gflg
‑
poly d
‑
galactosamine
‑
s
‑
hexyl
‑
ppa(bshp)按如下方法制备:500mg b
‑
gala
‑
sh溶于5ml ro h2o后加入10ml dmso配制成b
‑
gala
‑
sh/dmso混合液,80mg maleimide
‑
hexyl
‑
ppa溶于2ml dmso后搅拌下滴加到b
‑
gala
‑
sh/dmso混合液中得到墨绿色溶液,于室温环境避光反应过夜后透析(避光,mwco 3.5kda)2天除去dmso,离心(7000rpm
×
5min)后取上清液经水相滤头(0.45μm)过滤,滤液冻干得黑绿色固体粗品(520mg,产率89.7%)。将粗品用2ml水溶解后滴加到200ml乙腈中出现大量沉淀,离心(9000rpm
×
5min)处理后收集固体残留物并重复处理纯化三次,得到的固体再次溶于20ml去离子水后冻干得产物(bshp,450mg)。聚合物branched
‑
gflg
‑
poly d
‑
galactosamine
‑
s
‑
hexyl
‑
ppa(bshp)的氢谱如图8所示。
[0117]
对照例2、线性糖基聚合物光动力治疗体系的制备
[0118]
线性糖基聚合物光动力治疗体系的合成路线如图9所示:
[0119]
作为对照,设计合成了线性糖基聚合物光动力治疗体系。单体ma
‑
d
‑
galactosamine(722mg,2.31mmol)、ma
‑
pyss(50mg,0.20mmol)、链转移剂4
‑
氰基
‑4‑
(硫代苯甲酰)戊酸(cta,5.3mg,19.0μmol)与引发剂va044(2.1mg,6.5μmol)溶于h2o/ch3oh(3.5ml,1:4,v/v)的混合溶液中,将聚合瓶避光置于冰浴中并通氩气鼓泡除氧45分钟后将其密闭,避光置于45℃油浴反应12小时,用液氮淬灭反应后将反应液低温透析冻干,得淡红色固体linear
‑
poly d
‑
galactosamine
‑
pyss(l
‑
gala
‑
pyss:725mg,产率93.3%)。聚合物前体linear
‑
poly d
‑
galactosamine
‑
pyss的氢谱如图10所示。
[0120]
将620mg l
‑
gala
‑
pyss溶于10ml水溶液,加入600mg dtt处理过夜后透析(4℃,mwco 3.5kda)2天,样品用水相滤头处理后冻干得到白色固体粉末linear
‑
poly d
‑
galactosamine
‑
sh(l
‑
gala
‑
sh,560mg,产率93.3%)。聚合物前体linear
‑
poly d
‑
galactosamine
‑
sh的氢谱如图11所示。
[0121]
linear
‑
poly d
‑
galactosamine
‑
s
‑
ppa(lsp)的制备与可降解的支化/交联糖基聚合物光动力治疗体系的制备方法类似:500mg l
‑
gala
‑
sh溶于5ml ro h2o后加入10ml dmso配制成l
‑
gala
‑
sh/dmso混合液,80mg maleimide
‑
ppa溶于2ml dmso后搅拌下滴加到l
‑
gala
‑
sh/dmso混合液中得到的墨绿色溶液,于室温环境避光反应过夜后透析(避光,mwco 3.5kda)2天除去dmso,离心(7000rpm
×
5min)后取上清液经水相滤头(0.45μm)过滤,滤液冻干得黑绿色固体粗品(512mg,产率88.3%)。将粗品用2ml水溶解后滴加到200ml乙腈中出现大量沉淀,离心(10000rpm
×
5min)处理后收集固体残留物并重复处理纯化三次,得到的固体再次溶于20ml去离子水后冻干得产物(lsp,496mg)。聚合物linear
‑
poly d
‑
galactosamine
‑
s
‑
ppa(lsp)的氢谱如图12所示。
[0122]
对照例3、基于hpma的可降解支化/交联聚合物光动力治疗体系的制备
[0123]
作为糖基材料的对照,设计合成了基于hpma的可降解支化/交联聚合物光动力治疗体系。基于hpma的可降解支化/交联聚合物光动力治疗体系的合成路线如图13所示。具体合成方法如下:
[0124]
单体hpma(722mg,5.04mmol)、ma
‑
pyss(50mg,0.20mmol)、交联剂ma
‑
gflgkglfg
‑
ma(11.8mg,11.4μmol)、链转移剂ma
‑
gflgk
‑
cta(26.5mg,31.2μmol)与引发剂va044(3.4mg,10.4μmol)溶于h2o/ch3oh(3.6ml,1:4)的混合溶液中,将聚合瓶避光置于冰浴中并通氩气鼓泡除氧45分钟后将其密闭,避光置于45℃油浴反应12小时,用液氮淬灭反应后将反应液低温透析冻干,得淡红色固体branched
‑
gflg
‑
poly hpma
‑
pyss(b
‑
hpma
‑
pyss,740mg,产率91.2%)。聚合物前体branched
‑
gflg
‑
poly hpma
‑
pyss的氢谱如图14所示。
[0125]
将640mg b
‑
hpma
‑
pyss溶于10ml水溶液中,加入1000mgdtt处理过夜后透析(4℃,mwco 3.5kda)2天,样品用水相滤头处理后冻干得到白色固体粉末branched
‑
gflg
‑
poly hpma
‑
sh(b
‑
hpma
‑
sh,600mg,产率93.8%)。聚合物前体branched
‑
gflg
‑
poly hpma
‑
sh的氢谱如图15所示。
[0126]
branched
‑
gflg
‑
poly
‑
hpma
‑
s
‑
ppa(bhsp)的制备如下:500mg b
‑
hpma
‑
sh溶于5ml ro h2o后加入10ml dmso配制成b
‑
hpma
‑
sh/dmso混合液,80mg maleimide
‑
hexyl
‑
ppa溶于2ml dmso后搅拌下滴加到b
‑
hpma
‑
sh/dmso混合液中得到的墨绿色溶液,于室温环境避光反应过夜后透析(避光,mwco 3.5kda)2天除去dmso,离心(7000rpm
×
5min)后取上清液经水相滤头(0.45μm)过滤,滤液冻干得黑绿色固体粗品(500mg,产率86.2%)。将粗品用2ml水溶解后滴加到200ml丙酮中出现大量沉淀,离心(9000rpm
×
5min)处理后收集固体残留物并重复处理纯化三次,得到的固体再次溶于20ml去离子水后冻干得产物(bhsp,460mg)。聚合物branched
‑
gflg
‑
poly hpma
‑
s
‑
ppa(bhsp)的氢谱如图16所示。
[0127]
以下通过具体试验例证明本发明的有益效果:
[0128]
试验例1、聚合物的表征
[0129]
1、聚合物的粒径与电位测定
[0130]
称取bsp、bshp、lsp,纯水溶解稀释至终浓度为1.0mg/ml,使用粒度仪表征样品粒径和zata电位(每次测量重复三次),最终数据使用graphpad prism软件进行分析。同时将浓度为200μg/ml的样品滴加到铜网上自然风干,使用透射电镜观察粒径大小与形貌。
[0131]
通过透射电镜观察到各组材料的形貌如图17所示。bshp的组装体呈十分均匀的圆形纳米粒;相较而言bsp形成的纳米粒则并不是太规则,但也能观察到类似圆形的形貌;而不具有支化/交联结构的lsp则呈松散的丝状分布,没有聚集形成特殊形貌。
[0132]
如图18所示:通过dls测得bsp、bshp和lsp的粒径分别约为238nm、164nm和374nm,即bshp形成的粒子最为致密,bsp却相对松散但仍保持纳米粒子形态。三组材料的zeta电位
分别为
‑
16.81mv、
‑
19.23mv和
‑
21.97mv,均为电负性。
[0133]
2、聚合物的体外ros产生测定
[0134]
将bsp、bshp和lsp溶解稀释至光敏剂焦脱镁叶绿酸
‑
a(pyropheophorbide a,ppa)浓度为5.0μg/ml,同时加入sosg检测试剂至终浓度为2.5μm。吸取各组溶液100μl至96孔板,并设3个复孔,随后用660nm激光进行辐照(功率密度:10mw/cm2,持续时间:15min)。在0.5、1、1.5、2、3、5、10、15min时间点用多功能酶标仪测定荧光强度(激发波长为490nm,发射波长为525nm)并记录各样品在525nm波长处的荧光强度。用pbs作为空白对照。
[0135]
ppa能吸收特定波长光子而从基态跃迁到激发态,随后将吸收的能量转移到附近的氧气分子产生单线态氧。ppa经光照激发产生的ros可以通过试剂盒检测。其中,单线态氧荧光探针(singlet oxygen sensor green,sosg)是一种对单线态氧具有高度选择性的检测试剂,该指示剂最初具有弱蓝色荧光,与单线态氧作用后会发出与荧光素相似的绿色荧光(最大激发/发射波长约为504/525nm),绿色荧光强弱与产生的ros量成正相关。如图19所示,在吸收660nm激发能量后,bsp所产生的荧光强度最强,lsp次之,bshp最弱,且bsp荧光强度显著高于lps和bshp。说明bsp体外ros产生的量和效率更高,而bshp和lsp的ros产生效率则相对较低。进一步说明bsp对肿瘤有更好的杀伤作用。
[0136]
3、聚合物体外ic50值测定
[0137]
将4t1细胞按5
×
104个/每孔的细胞浓度接种于96孔板中,待细胞贴壁(约孵育12小时)后吸弃培养基,加入用培养基配制的含bsp、lsp和bshp的梯度浓度(ppa浓度:50、20、10、5、2、1、0.5、0.1、0.01μg/ml)溶液后继续孵育6小时,孵育完成后分别给予2j/cm2剂量光照,再孵育12小时后根据试剂盒说明书使用cck8试剂,通过酶标仪检测450nm左右吸光度。使用graphpad prism(vison 8.0)软件对测定结果进行作图分析。
[0138]
通过体外细胞实验进行了ic
50
值的比较(图20)。在同样的ppa浓度梯度、光照剂量以及培养条件下,三组聚合物ic50值分别为bsp 0.25μg/ml,lsp 0.91μg/ml,bshp 4.8μg/ml,说明bsp对肿瘤细胞杀伤效果最强。
[0139]
4、聚合物体内肿瘤部位信号检测
[0140]1×
106个4t1细胞接种于balb/c小鼠(20克,6
‑
8周龄)侧腹部,待肿瘤生长到直径约5mm左右开始进行实验。将小鼠随机分为3组(每组5只),分别经尾静脉注射ppa浓度为5mg/kg的不同组材料(bsp、bshp和lsp)。于注射完成不同时间点(5min、30min、1h、3h、6h、12h和24h)通过活体荧光成像系统观察肿瘤部位荧光信号,并通过数据分析系统处理数据并分析。
[0141]
结果表明,bsp材料在注射后肿瘤部位荧光强度逐渐升高,6小时左右达到最高,并且在各个时间点的肿瘤部位荧光强度均远高于lsp和bshp组(图21)。
[0142]
在同样ppa浓度下,bsp无论是体外ros的产生量、对肿瘤细胞的毒性作用和在肿瘤组织聚集度均高于lsp和bshp,这保证了光动力治疗的效果。bsp可作为光敏剂,用于光动力治疗肿瘤具有优异的效果。
[0143]
试验例2、聚合物包载奥拉帕尼及其表征
[0144]
1、聚合物包载奥拉帕尼
[0145]
聚合物包载奥拉帕尼通过透析法制备,称取300mg lsp、bsp、bshp及bhsp分别溶于5ml去离子水,并加入10ml dmso,得到含有材料的h2o/dmso混合溶液。30mg奥拉帕尼
(olaparib)溶于2ml dmso后滴加到含有材料的h2o/dmso混合溶液。避光搅拌过夜,将反应液透析(3.5kda mwco)2天去除dmso溶剂,然后用水相滤膜处理后冻干,得到包载奥拉帕尼的聚合物,聚合物依次命名为lspo、bspo、bshpo和bhspo(o代表olaparib)。
[0146]
2、奥拉帕尼包载量的测定
[0147]
lsp、bsp、bshp和bhsp对于奥拉帕尼的包载量通过hplc结果计算。载药量(wt%)=(包载的药物的质量/聚合物与包载药物的总质量)
×
100%。将各组材料分别用流动相溶解后超声,并将有机相滤头处理后的滤液进行hplc检测。色谱柱型号为(column name:shim
‑
pack glst,c18,5μm.size:250
×
4.6mm i.d.),测试柱温为30℃,流速为1.0ml/min,进样量为20μl,样品浓度约为100μg/ml,检测器选择为uv light(276nm),流动相为ch3oh/h2o(v:v=1:1)的混合溶液。
[0148]
结果如表2所示。bsp聚合物的包载量最高,达到4.03%;bshp的包载量为2.17%,差于bsp。相较而言,对照材料lsp和bhsp无法有效包载小分子药物,其包载量分别仅为0.33%和0.13%。说明bsp“合适”的组装堆叠形态能够包载更多的小分子药物。通过hplc收集包载的小分子药物进行检测,hrms结果表明olaparib仍然为活性原药(图22)。
[0149]
表2.各组聚合物的ppa含量及对应olaparib的包载量
[0150][0151]
3、bspo的相关表征
[0152]
用dls测量包载奥拉帕尼的聚合物bspo(1.0mg/ml)的水相粒径和zeta电位,并进一步通过tem考察其形貌(制样浓度为200μg/ml)。用紫外
‑
可见分光光度计在200
‑
800nm范围内检测样品bsp、bspo、ppa和olaparib的紫外光谱,通过荧光光谱仪测定bsp、bspo、ppa和olaparib的荧光光谱。通过sosg检测试剂评价bsp和bspo的体外单线态氧生成效率的变化,具体方法与试验例1体外ros产生测定的条件方法相同。
[0153]
如图23所示,dls结果表明bspo水相粒径约为178nm,其表面同样为负电荷;而tem结果表明其形成了尺寸相对均一的纳米粒子。这表明相较于bsp,包载了小分子奥拉帕尼后的bspo结构可能更为致密。
[0154]
bspo的荧光光谱性质:如图24所示,相同ppa浓度下,bsp与bspo在680nm左右的荧光强度无明显差别,说明包载奥拉帕尼后并不影响ppa的荧光性质。
[0155]
与此同时,在紫外光谱图中(图25)bsp和bspo在330
‑
800nm左右的紫外光谱基本重叠,说明紫外光谱在这一波长范围内没有改变;而在300nm左右,紫外光谱略有区别,即bspo在300nm左右的光谱发生了变化。奥拉帕尼的紫外吸收峰在276nm左右,这一变化是ppa的紫外峰与奥拉帕尼的紫外峰重叠引起的,这一结果一定程度上说明了bsp能够有效包载奥拉帕尼,且包载奥拉帕尼不会影响聚合物产生ros能力。
[0156]
如图26所示,bspo与bsp相比,其体外的ros产生量和效率没有明显差异,说明包载
奥拉帕尼后仍能高效产生ros。
[0157]
上述试验结果说明:糖基聚合物bsp可以作为光敏剂,用于光动力治疗,其在肿瘤部位有良好的聚集效果,并且对肿瘤有良好的杀伤作用;同时,糖基聚合物bsp可以作为药物载体包裹药物,如奥拉帕尼,进一步用于疾病治疗。其包裹药物的载药量高,作为给药系统效果优异。
[0158]
本发明合成了不同结构的糖基聚合物载体bsp、bshp和lsp,以及基于phpma的药物载体bshp。实验结果表明在相同光敏剂焦脱镁叶绿酸
‑
a(pyropheophorbide a,ppa)浓度的情况下,相比于bshp和lsp,bsp光照后ros的产生效率最高。肿瘤细胞毒性实验发现bsp、bshp和lsp三种聚合物载体的半抑制浓度(half maximal inhibitory concentration,ic50)分别为0.25μg/ml、4.8μg/ml和0.91μg/ml,bsp具有更明显的肿瘤细胞杀伤作用。
[0159]
本发明还制备了包载奥拉帕尼的双药递送系统bspo。包载药物奥拉帕尼能力的研究发现,bsp对药物的包载能力最强,作为药物载体最优。
[0160]
可见本发明制备的糖基聚合物载体bsp在肿瘤部位的聚集浓度高,具有良好的光动力治疗效果,对肿瘤杀伤作用强,是一种良好的光敏剂;且作为药物载体包载量大,是一种优异的给药系统。
[0161]
综上,本发明提供了一种糖基聚合物bsp,该糖基聚合物克服了现有pdt光敏剂肿瘤部位聚集不够,对肿瘤杀伤作用不强的缺点,可以在肿瘤部位聚集,具有良好的靶向作用;其具有光动力效应,作为光敏剂用于光动力治疗ros产生效率高,对肿瘤细胞杀伤作用强。同时,该糖基聚合物还可以作为给药系统包载药物,特别是肿瘤药物,如奥拉帕尼,用于肿瘤治疗,效果优异。本发明糖基聚合物用于制备治疗肿瘤的药物具有优良的应用前景。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。