抗生素治疗剂及其用途
发明领域
1.本发明涉及抗生素治疗剂(antibiotic therapeutics)的组合物及其用途。
2.发明背景
3.可生物降解的药物递送系统是有利的,因为它们避免了对用于去除不可降解的药物耗尽装置(drug depleted device)的另外的医疗干预的需要。这些聚合物及其降解组分必须具有包括以下的若干属性:与生物组织的相容性、可忽略的毒性和易于从身体中清除。可生物降解的聚合物通常是疏水性的,从而在施用之后在生理环境中保持其完整性。
4.已经开发了包含抗生素诸如庆大霉素的可生物降解的系统。然而,它们经常提供抗生素的不恒定的释放。此外,这些系统中的一些已经被报告引起局部的超敏反应(hypersensitivity reaction)。
5.先前的体外和体内研究已经表明,由蓖麻油酸和癸二酸形成的聚(酯-酸酐)可以作为方便并且安全的可生物降解聚合物用于药物的局部施用。还针对在骨髓炎的治疗中的庆大霉素施用,专门评估了这些共聚物[1],示出良好的耐受性、有利的局部释放动力学以及没有炎性不良反应的迹象。
[0006]
wo 2016/097848[2]公开了被表征为交替或半交替的酯键和酸酐键的共聚物、用于其生产的方法及其用途,特别是作为用于药物递送的载体的用途。该共聚物被表征为可再现的产品规格,包括受控的黏度和分子量,并且示出在室温持续数月是稳定的。
[0007]
wo 2018/178963[3]公开了包含至少一种抗生素和可生物降解的聚(酯-酸酐)的储库系统(depot system),以在注射部位处提供抗生素的延长的局部释放,同时将全身抗生素水平维持在亚治疗浓度(sub-therapeutic concentration)。
[0008]
虽然用于局部递送抗生素的可生物降解系统克服了现有的不可生物降解的局部治疗的缺点中的很多缺点,但是它们可能不足以完全地根除在例如骨形成和牙齿相关的感染中涉及的细菌。因此,需要治疗方式(therapeutic modality)的另外的进步。
[0009]
由于聚酸酐的表面侵蚀性质,它们已经作为用于若干种药物的受控递送(controlled delivery)的载体被研究。聚酸酐对水具有固有的高反应性,这促使快速的水解降解。由于高速率的水解,聚酸酐经受表面侵蚀而不是整体降解。基于聚酸酐的颗粒已经在许多制剂中被广泛研究用于有效药物递送。然而,与聚酯相比,市场上存在的聚酸酐产品的数量较少。虽然聚酸酐的合成和放大生产是容易并且便宜的,但它们表现出短的储存期。聚酸酐在储存期间易于经由酸酐交换进行水解降解和解聚,并且因此可能连同分解产物一起产生。因此,聚酸酐需要保存在冷冻储存条件,这限制了它们在药物递送产品中的使用。因此,聚酸酐产品在医疗领域(例如药物的载体)中的可用性不太有吸引力。一个这样的实例是在[4-6]中报告的基于蓖麻油酸和癸二酸的聚(酯-酸酐)。
[0010]
参考文献
[0011]
[1]brin等人,2009,j biomater sci polym ed,20,1081-1090;krasko等人,2007j.control release,117,90-96;
[0012]
[2]wo 2016/097848;
[0013]
[3]wo 2018/178963;
[0014]
[4]us 10,774,176;
[0015]
[5]us 2020/0101163;
[0016]
[6]domb等人,2017,j of controlled release,257,156-162。
[0017]
发明概述
[0018]
本文公开的本发明涉及独特的基于可生物降解和生物相容性的聚合物的组合物,用于递送无限种类的抗生素。本发明的制剂可以被注射或嵌入组织中用于获得最大效果,或者甚至可以局部应用以用于获得局部的非全身效果。该递送系统提供了抗生素药物的高的局部浓度,从而不仅实现了对现有的状况或感染的治疗,而且还在延长的时间段内预防了感染的再形成。
[0019]
本发明的制剂通常基于聚酸酐,该聚酸酐表现出相对先前在本领域中公开的那些改善的性质。聚酸酐是由癸二酸(sa)和蓖麻油酸(ra)构建的窄多分散性的聚合物,其通过在采用每个羧酸基团1摩尔当量或更少的乙酸酐的情况下并且在不存在溶剂的情况下sa和ra的熔融缩合(melt condensation)来制备。聚酸酐具有形式-(sa-ra)n-,其中sa是癸二酸并且ra是蓖麻油酸,并且其中n是在10和100之间的整数。这种聚酸酐在本文被称为本发明的聚合物或者本发明的载体。
[0020]
溶剂的不存在和多种前体的依次添加允许产生这样的最终产物,该最终产物被良好地表征并且是可再现的,以满足最高标准的监管要求,并且表现出窄多分散性。术语“窄多分散性或其任何语言变型当参照本发明的聚合物作出时,定义了具有基本上相同的组成(重复基团的类型和重复的方式)和分子量的材料的集合。由比率mw/mn(其中mw是重均分子量并且mn是数均分子量)定义的本发明的聚合物的窄多分散性低于2.5或低于2。换言之,本发明的窄分散性聚合物或窄多分散性聚合物具有不大于2.5或2(或在2.5和1之间的值,或在2和1之间的值)的多分散性值。
[0021]
本发明的聚合物还表现出高再现性,即与聚合物平均分子量不大于30%的偏差的聚合物分子量的再现性。
[0022]
术语“在不存在溶剂的情况下”在本文指的是本发明的工艺作为不具有溶剂或具有微量的溶剂的性质,所述溶剂可以源自与前体材料一起存在的杂质。这样的杂质不超过所用的反应材料的总重量的0.001%、0.005%、0.01%、0.05%或0.1%(w/w)。
[0023]
本发明的聚合物通过包括以下的工艺制备:
[0024]-使癸二酸(sa)和蓖麻油酸(ra)在允许sa酯化(以获得sa的单酯或其二酯或其混合物)的条件下反应;以及
[0025]-将(单或二或其混合物)酯化的sa转化为窄多分散性聚酸酐。
[0026]
本发明的工艺允许在本体中(在熔体中)直接缩合,而无需预反应以形成所用的任何材料前体的聚合物或低聚物。在示例性的工艺中,癸二酸(sa)(二羧酸)与蓖麻油酸(ra)(羟基-链烷酸)以30:70w/w的比反应,以形成sa-ra二聚体和ra-sa-ra三聚体的混合物,在反应产物中具有极少或没有ra或ra-ra酯分子。此后,用每个游离羧酸基团不大于1摩尔当量的乙酸酐(通常是2个游离羧酸基团,并且因此不大2摩尔当量)处理sa-ra和ra-sa-ra混合物(不含前体分子和ra-ra分子),以使游离酯乙酰化,并且然后使乙酰化的链段聚合成具有重复的
…
ra-sa-ra-sa
…
序列的窄分散性聚酸酐。该工艺在图1中描述。
[0027]
sa和ra的二聚体和三聚体的混合物可以被用于形成非均相聚合物(heterogeneous polymer),该非均相聚合物由在sa和ra之间的酸酐键和酯键以及在两个ra单元之间极少的酯键组成。在另一个方面中,sa单体沿聚合物链的酸酐二联体(anhydride diad)的形成可能限制聚合物的储存稳定性。因此,在本发明的工艺中,sa和ra之间的摩尔比通常是当量的或有利于ra的。换句话说,ra的量优选地等于sa的量或是sa的量的2倍(1:1至1:2摩尔当量)。在一些实施方案中,sa:ra重量比分别是1:1、1:1.1、1:1.2、1:1.3、1:1.4、1:1.5、1:1.6、1:1.7、1:1.8、1:1.9、1:2。
[0028]
在一些实施方案中,sa:ra之间的摩尔比在1:1和1:2之间的范围内,以避免在ra单元之间的酯键形成,使得聚合物仅在sa和ra之间包含酸酐键和酯键。
[0029]
在一些实施方案中,对于sa结构单元和ra结构单元,重量比分别是30:70、35:65或25:75。
[0030]
过量的ra允许sa的单酯化和二酯化(具有一定量的单酯化形式),并且避免ra的酯二聚体的形成。sa-ra和sa-ra-sa混合物(本文中为“二聚体-三聚体混合物”)通过在高于80℃的温度加热以所示比的sa和ra的混合物来获得。在一些实施方案中,温度在80℃和200℃之间、在100℃和190℃之间、在100℃和180℃之间、在100℃和170℃之间、在100℃和160℃之间、在100℃和150℃之间、在100℃和140℃之间、在100℃和130℃或在100℃和120℃之间。
[0031]
两种组分的缩合包括直接酯缩合以提供二聚体-三聚体二羧酸低聚物混合物。二聚体-三聚体低聚物通过用乙酸酐活化羧酸端部而被聚合成聚酸酐。所用的乙酸酐的量不大于低聚物中每一个游离羧酸基团一摩尔当量的乙酸酐。二聚体sa-ra具有两个游离羧酸基团。类似地,三聚体sa-ra-sa具有2个游离羧酸基团。因此,可以使用不大于2摩尔当量的乙酸酐。在一些实施方案中,乙酸酐的量是2摩尔当量、1.9摩尔当量、1.8摩尔当量、1.7摩尔当量、1.6摩尔当量、1.5摩尔当量、1.4摩尔当量或1.3摩尔当量。
[0032]
在一些实施方案中,乙酰化步骤可以在高于40℃的温度进行。在一些实施方案中,乙酰化温度在40℃和乙酸酐的沸点之间。在一些实施方案中,乙酰化温度在40℃和90℃之间、在40℃和100℃之间、在40℃和110℃之间、在80℃和酰化酸酐的沸点之间。用于低聚物的酰化-活化的温度随时间变化而变化,反应时间越长,待应用的温度越低。可以在压力下使二酸低聚物与乙酸酐反应以加速反应,或者在微波加热下进行反应。这些方法要求调整反应条件,使得低聚物被乙酰化并且不劣化。此外,可以应用其他乙酰化方法,包括乙酰氯与除酸剂的反应。
[0033]
可以在乙酰化后升高温度以使乙酰化的前体缩合,以形成前述的二聚体/三聚体混合物。
[0034]
通过聚合实现向本发明的窄多分散性聚合物的转化。将二聚体-三聚体混合物聚合成本发明的聚合物可以通过在低压和升高的温度下加热乙酰化的二聚体和三聚体来实现。在一些实施方案中,聚合在真空和加热中可实现。热条件可以包括将乙酰化的二聚体-三聚体混合物加热至在100℃和200℃之间、在100℃和190℃之间、在100℃和180℃之间、在130℃和170℃之间、在130℃和160℃之间、在130℃和150℃之间或在130℃和140℃之间的温度。在一些实施方案中,温度在120℃和170℃之间或在130℃和160℃之间。反应时间是重要的参数,因为反应温度越高,反应时间越短。存在形成低聚物和聚合物所需的最短时间;
较长的反应时间对低聚物组成或聚合物分子量没有影响或具有很小的影响。反应时间取决于批量大小和反应条件,包括混合方法和速率以及所应用的真空概况(vacuum profile)。
[0035]
在一些实施方案中,聚合在如所指定的高热条件、在真空下可实现。
[0036]
在一些实施方案中,该工艺包括:
[0037]-在80℃和200℃之间的温度使sa和ra反应,以获得sa的单酯(sa-ra)和二酯(sa-ra-sa)的混合物;以及
[0038]-在允许单酯和二酯聚合成聚酸酐的条件下使混合物与乙酸酐反应。
[0039]
在一些实施方案中,该工艺包括:
[0040]-在80℃和200℃之间的温度使sa和ra反应,以获得sa的单酯(sa-ra)和二酯(sa-ra-sa)的混合物;以及
[0041]-使混合物与乙酸酐反应,以使单酯和二酯的混合物乙酰化;以及
[0042]-在允许聚合成聚酸酐的条件下热处理乙酰化的混合物。
[0043]
在一些实施方案中,该工艺包括:
[0044]-在存在乙酸酐的情况下,使sa和ra在80℃和200℃之间的温度反应,以获得如本文的sa的单酯和二酯的混合物;以及
[0045]-在真空中在100℃和200℃之间的温度热处理乙酰化的混合物,允许聚合以提供聚酸酐。
[0046]
因此,本发明的聚合物是式-(sa-ra)n-的窄多分散性聚酸酐,其中sa是癸二酸并且ra是蓖麻油酸,并且其中n是在10和100之间的整数,所述窄多分散性聚酸酐具有低于2.5或低于2的mw/mn值(其中mw是重均分子量并且mn是数均分子量)或在1和2.5之间或1和2之间的值,通过如上文公开的工艺制备,其中混合物或二聚体和三聚体二羧酸通过酸酐键连接以形成链。本发明的工艺不包括此类生产多分散性聚酸酐的工艺。本发明的工艺不含形成或利用衍生自sa(由sa组成)或衍生自ra(由ra组成)的聚合物或低聚物的步骤。被排除在本发明的范围之外的一种这样的工艺是利用sa和ra的工艺,并且在出版物[4-6]中公开。本发明的聚合物是共同未决的美国专利申请第63/062,563号和要求其优先权的任何共同未决的申请的主题,其中的每一个申请在本文中通过引用并入。
[0047]
因此,在其所有实施方案中的载体通过如本文的方法或工艺被制备,其中方法或工艺或制备不包括聚癸二酸的使用。
[0048]
高度可再现的批次间聚合物分子量提供了改进的可再现的黏度,这允许可预测的可注射性(injectability)、高度可再现的组合物和药物释放概况,连同可预测的、可管控的、具有窄标准偏差的聚合物降解速率,以及高纯度(极少或没有乙酸和乙酸酐分子的反应物杂质),本发明的聚合物优于本领域中讨论的聚合物。因此,本发明的聚酸酐在医疗领域中的可用性,例如作为药物载体,为新一代的药物载体打开了大门。
[0049]
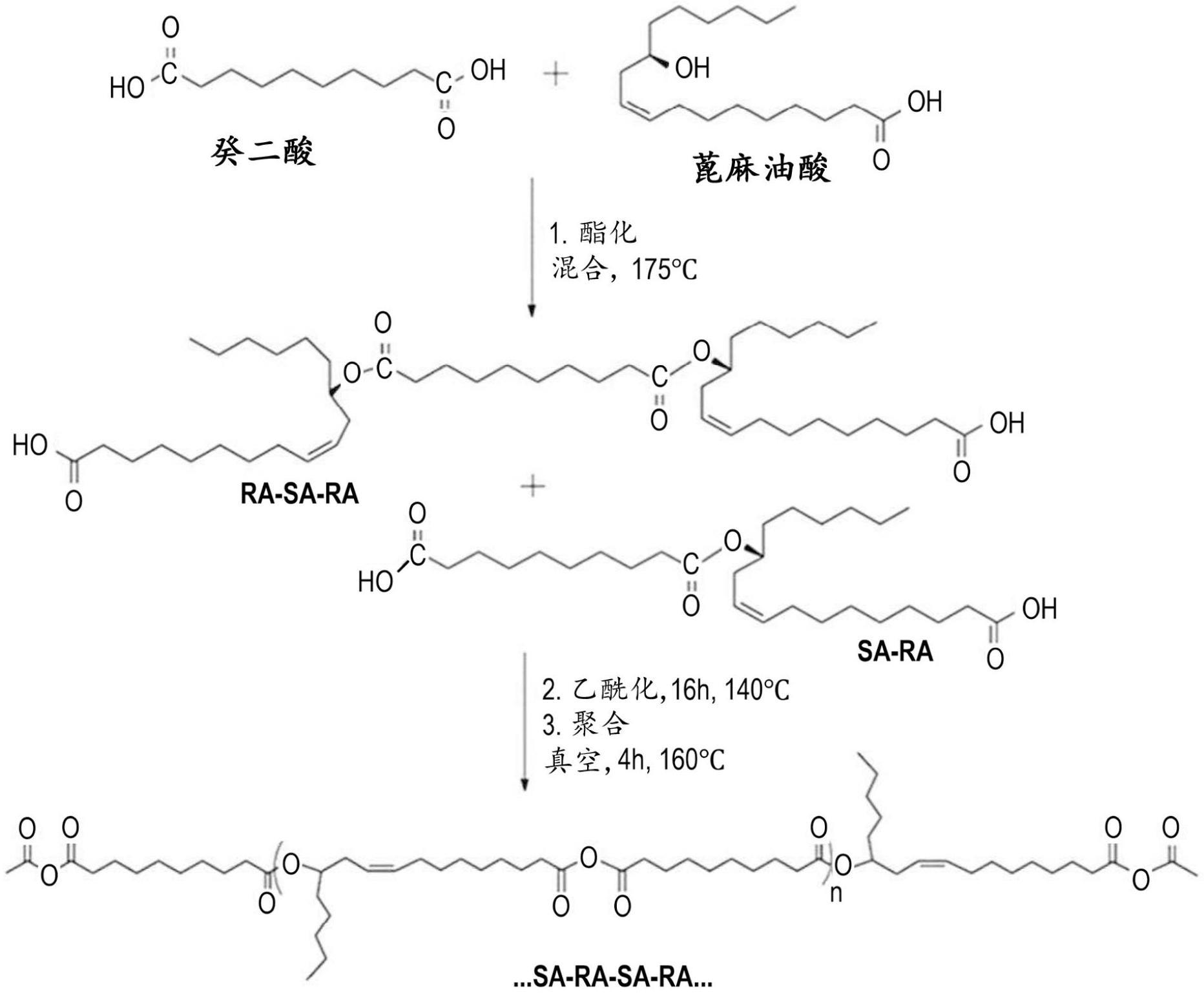
因此,在第一方面中,提供了包含本发明的聚合物(如所定义的或如所制备的)和至少一种抗生素剂的抗生素制剂或抗微生物制剂。
[0050]
更具体地,本发明的制剂包含至少一种抗生素剂和呈聚酸酐的形式的载体,所述聚酸酐包含癸二酸(sa)和蓖麻油酸(ra),所述载体具有在1和2.5之间的mw/mn值。载体是式-(sa-ra)n-的聚酸酐,其中n是在10和100之间的整数。如本文所提及的,聚酸酐通过以下来制备:a.sa和ra的熔融缩合以形成二羧酸低聚物;b.采用乙酸酐的低聚物活化;c.熔融缩
聚以形成聚酸酐。在不存在溶剂的情况下,在存在每个羧酸基团1摩尔当量或更少的乙酸酐的情况下可实现低聚物活化。
[0051]
如本文使用的,抗生素“制剂”或抗微生物“制剂”是包含至少一种抗生素剂和载体的药物级制剂或药物级组合物,所述载体包含本发明的聚合物或由本发明的聚合物组成。在将要修改本发明的制剂的性质的情况下,在一些实施方案中,除了本发明的聚合物之外,所用的载体还可以包括其他可接受的载体,诸如例如媒介物、辅料、赋形剂或稀释剂。除了本发明的聚合物之外,使用另外的载体的选择将部分地取决于特定的抗生素剂,以及用于施用制剂的特定方法和制剂的特定形式。
[0052]
在一些实施方案中,抗生素制剂/抗微生物制剂包含抗生素剂和呈式-(sa-ra)n-的聚酸酐的形式的载体,其中sa是癸二酸并且ra是蓖麻油酸,并且其中n是在10和100之间的整数,所述聚酸酐具有低于2.5或低于2的mw/mn值(其中mw是重均分子量并且mn是数均分子量)或在1和2.5之间或1和2之间的值。
[0053]
在一些实施方案中,聚酸酐通过采用每个羧酸基团1摩尔当量或更少的乙酸酐、在不存在溶剂的情况下sa和ra的熔融缩合来制备。换句话说,聚酸酐不是通过涉及溶剂的使用或单独的ra或sa的聚合的工艺制备的。
[0054]
本发明还提供了呈式-(sa-ra)n-的聚酸酐的形式的载体用于制备包含至少一种抗生素剂的抗生素制剂的用途,其中sa是癸二酸并且ra是蓖麻油酸,并且其中n是在10和100之间的整数,所述聚酸酐具有低于2.5或低于2的mw/mn值(其中mw是重均分子量并且mn是数均分子量)或在1和2.5之间或1和2之间的值。
[0055]
此外,提供了抗生素剂,其用于制备包含抗生素剂和呈式-(sa-ra)n-的聚酸酐的形式的载体的抗生素制剂,其中sa是癸二酸并且ra是蓖麻油酸,并且其中n是在10和100之间的整数,所述聚酸酐具有低于2.5或低于2的mw/mn值(其中mw是重均分子量并且mn是数均分子量)或在1和2.5之间或1和2之间的值。
[0056]
包含抗生素剂和本发明的聚合物的本发明的制剂可以通过多种方式来形成。在一些情况下,制剂通过将如所定义的本发明的聚合物与至少一种抗生素剂混合来形成。在这样的情况下,将可测量剂量量的抗生素剂与适当量的聚合物混合,以获得均质制剂。在其他情况下,制剂通过在聚合物的制备期间将抗生素剂与聚合物前体混合来形成。
[0057]
一般而言,本发明的制剂可以被配置为受控释放制剂。术语“受控递送”在本文中以其最广泛的意义被使用,以指示一种制剂,由此调节抗生素剂从制剂中的排出和剂穿过组织的渗透、其在组织和血液循环中的可及性(accessibility)和生物利用度和/或靶向特定的作用组织,以随着时间的推移实现特定的效果。其包括抗生素剂的立即递送、延长递送和持续递送、针对降解的药物保护、优先代谢、清除或递送至特定组织。在本发明的制剂中包含的抗生素剂的受控释放可以通过如本领域中已知的若干种手段来获得。
[0058]
典型地,本发明的制剂被配置为延长递送制剂或持续递送制剂。
[0059]
术语“延长递送”意味着抗生素剂从制剂以及进入组织中延缓的渗透和/或释放。换句话说,在延长递送中,在延迟期之后,并且在这种情况下,在施用之后至少约10min、20min、30min、40min、50min、60min、70min、80min、90min、100min、110min、120min、130min、140min、150min、160min、170min、180min之后以及另外在至少约3h、4h、5h、6h、7h、8h、9h、10h或更长时间之后,可以在组织或循环中检测到或测量到剂。延长递送还适用于在施用之
后另外延迟至少约10min、20min、30min、40min、50min、60min、70min、80min、90min、100min、110min、120min、130min、140min、150min、160min、170min、180min以及另外在至少约3h、4h、5h、6h、7h、8h、9h、10h或更长时间之后靶向器官和组织。
[0060]
术语“持续递送”意味着剂从制剂中以及进入组织或循环中的连续释放和/或渗透的概况,或者换句话说,剂从制剂中以及进入组织或循环中的释放和/或渗透在施用之后至少约10min、20min、30min、40min、50min、60min、70min、80min、90min、100min、110min、120min、130min、140min、150min、160min、170min、180min之后以及另外在至少约3h、4h、5h、6h、7h、8h、9h、10h或更长时间之后达到平台状态或稳定状态,并且该平台状态或稳定状态持续至少约1h、2h、3h、4h、5h、6h、7h、8h、9h、10h、11h、12h、13h、14h、15h、16h、17h、17h、18h、19h、20h或更长时间。
[0061]“抗生素"剂是意图由人类或动物使用的药物,以用于抑制或破坏或预防微生物的感染,或治疗或预防由细菌介导或引起的疾病的发展。该术语不包括具有化学治疗活性的抗生素材料。根据本发明使用的抗生素剂是已知具有抗细菌活性或抗微生物活性的任何这样的剂。换言之,抗生素是被施用于受试者以实现对由细菌或某些寄生虫引起的感染的治疗或预防的任何这样的剂。在一些实施方案中,细菌是球菌属细菌(cocci bacteria)、芽孢杆菌属细菌(bacillus bacteria)、立克次氏体属细菌(rickettsia bacteria)、支原体属细菌(mycoplasma bacteria),以及其他。
[0062]
在一些实施方案中,细菌选自革兰氏阳性细菌和革兰氏阴性细菌。
[0063]
在一些实施方案中,选择抗生素剂来治疗或预防由革兰氏阳性细菌诸如链球菌属、葡萄球菌属和肉毒杆菌属引起的感染。在一些实施方案中,选择抗生素剂来治疗或预防由革兰氏阴性细菌诸如霍乱、淋病、大肠埃希杆菌(escherichia coli)(大肠杆菌(e.coli))、铜绿假单胞菌和鲍曼不动杆菌引起的感染。
[0064]
在一些实施方案中,抗生素剂被选择以治疗或预防由选自以下的细菌引起的感染:尿气球菌(aerococcus urinae)、沙眼衣原体、粪肠球菌、坏死梭杆菌、具核梭杆菌、卡他莫拉菌、淋病奈瑟菌、脑膜炎奈瑟菌(neisseria meningitides)、有害片球菌(pediococcus damnosus)、金黄色葡萄球菌、溶血性葡萄球菌、腐生葡萄球菌、无乳链球菌、牛链球菌、肺炎链球菌、化脓性链球菌、嗜水气单胞菌、溶血隐秘杆菌、炭疽杆菌、犬咬嗜二氧化碳菌(capnocytophaga canimorsus)、肺炎嗜衣原体(chlamydophila pneumoniae)、鹦鹉热嗜衣原体(chlamydophila psittaci)、肉毒杆菌、艰难梭菌、破伤风梭菌、白喉棒状杆菌、杰氏棒状杆菌、大肠埃希杆菌、产气克雷伯氏菌(klebsiella aerogenes)、嗜肺军团菌、单核细胞增生李斯特菌、麻风分枝杆菌、结核分枝杆菌(plesiomonas shigelloides)、类志贺邻单胞菌(plesiomonas shigelloides)、中间普雷沃菌、牙龈卟啉单胞菌(porphyromonas gingivalis)、产丙酸丙酸杆菌(propionibacteriumacidipropionici)、斯氏普罗威登斯菌(providencia stuartii)、鼠伤寒沙门氏菌、粘质沙雷氏菌、霍乱弧菌、创伤弧菌、亚麻短杆菌(brevibacterium linens)、小蛛立克次氏体(rickettsia akari)、康氏立克次氏体(rickettsia conorii)、猫立克次氏体(rickettsia felis)、普氏立克次氏体(rickettsia prowazekii)、立氏立克次氏体(rickettsia rickettsii)、斑疹伤寒立克次氏体(rickettsia typhi)、阿氏疏螺旋体(borrelia afzelii)、伯氏疏螺旋体(borrelia burgdorferi)、赫氏疏螺旋体(borrelia hermsii)、大肠弯曲杆菌(campylobacter coli)、
肝螺杆菌(helicobacter hepaticus)、幽门螺杆菌(helicobacter pylori)、问号钩端螺旋体、减少螺菌(spirillum minus)、梅毒螺旋体(treponema pallidum)、品他病密螺旋体(treponema carateum)、齿垢密螺旋体(treponema denticola)、发酵支原体(mycoplasma fermentans)、鸡毒支原体(mycoplasma gallisepticum)、生殖支原体、猫血支原体(mycoplasma haemofelis)、人支原体、猪肺炎支原体、未识支原体(mycoplasma incognitus)、穿透支原体(mycoplasma penetrans)、肺炎支原体,以及其他。
[0065]
在一些实施方案中,抗生素剂基于其治疗或预防由细菌介导或引起的疾病或状况的能力来选择。一般而言,细菌可以通过多种机制引起疾病:(1)通过分泌或排泄毒素,如在肉毒中毒中;(2)通过在内部产生毒素,该毒素当细菌瓦解时被释放,如在伤寒中;(3)或者通过诱导对其抗原性的敏感性,如在结核病中。还可以涉及其他机制。因此,疾病或状况可以是肉毒中毒、伤寒、结核病、霍乱、白喉、细菌性脑膜炎、破伤风、莱姆病、淋病和梅毒中的任何一种或更多种。
[0066]
抗生素剂可以选自青霉素类、四环素类、头孢菌素类、喹诺酮类、林可霉素类、大环内酯类、磺酰胺类、糖肽类、氨基糖苷类和碳青霉烯类。
[0067]
在一些实施方案中,抗生素剂是阿莫西林、氨苄西林、双氯西林、苯唑西林、青霉素v钾、去甲金霉素(demeclocycline)、多西环素(doxycycline)、伊拉瓦环素(eravacycline)、米诺环素(minocycline)、奥马环素(omadacycline)、四环素、头孢克洛、头孢地尼、头孢噻肟、头孢他啶、头孢曲松、头孢呋辛、环丙沙星、左氧氟沙星、莫西沙星、克林霉素(clindamycin)、林可霉素(lincomycin)、阿奇霉素(azithromycin)、克拉霉素(clarithromycin)、红霉素、达巴万星(dalbavancin)、奥利万星(oritavancin)、特拉万星(telavancin)、万古霉素、庆大霉素、妥布霉素(tobramycin)、阿米卡星(amikacin)、亚胺培南、西司他丁、美罗培南(meropenem)、多利培南(doripenem)、厄他培南(ertapenem),以及其他,或其药学上可接受的盐。
[0068]
在一些实施方案中,抗生素剂是以下中的至少一种:氨曲南、头孢呋辛、头孢氨苄、克林霉素、万古霉素、头孢他啶、头孢唑啉、头孢曲松、头孢菌素、哌拉西林、他唑巴坦、妥布霉素、左氧氟沙星、阿莫西林、克拉维酸和庆大霉素,或其药学上可接受的盐。
[0069]
在一些实施方案中,抗生素剂是头孢呋辛。
[0070]
在一些实施方案中,抗生素剂是氨基糖苷类。在一些实施方案中,氨基糖苷类抗生素是以下中的至少一种:卡那霉素a、阿米卡星、妥布霉素、地贝卡星、庆大霉素、西索米星、奈替米星、新霉素b、新霉素c或新霉素e和链霉素,或其药学上可接受的盐。
[0071]
在一些实施方案中,氨基糖苷类抗生素是庆大霉素或其药学上可接受的盐(例如硫酸庆大霉素)。
[0072]
在一些实施方案中,抗生素剂是以下中的至少一种:安普霉素、阿贝卡星、阿司米星、贝卡那霉素(bekanamycin)、二氢链霉素、依沙芦星(elsamitrucin)、磷霉素/妥布霉素、g418、潮霉素b、异帕米星、春日霉素、来霉素、利维霉素(lividomycin)、小诺霉素、新霉胺(neamine)、诺尔丝菌素(nourseothricin)、巴龙霉素、普拉佐米星(plazomicin)、核糖霉素、链双霉素(streptoduocin)、妥妥霉素(totomycin)和威大霉素(verdamicin),或其药学上可接受的盐。
[0073]
在一些实施方案中,抗生素剂是以下中的至少一种:氨苄西林、诺氟沙星、磺胺甲
噁唑(sulfamethoxazole)、氟甲喹和两性霉素b,或其药学上可接受的盐。
[0074]
通过本发明还提供了利用本发明的制剂的治疗或预防的方法。
[0075]
在一个方面中,提供了用于治疗或延缓或预防(例如由至少一种细菌介导的)感染性疾病或紊乱的进展的方法,该方法包括向受试者(人类或非人类)施用有效量的如本文描述的在本发明的制剂中的抗生素剂。
[0076]
如本文使用的术语“治疗”指的是施用治疗量的本发明的制剂,该制剂有效地缓解与疾病例如感染性疾病相关的不期望的症状,在这样的症状发生之前防止它们的表现,减缓疾病的进展(在本文还被称为“延缓进展”),减缓症状的恶化,增强缓解期的开始,减缓在疾病的进展性慢性期(progressive chronic stage)中引起的不可逆性损伤,延缓所述进展期的开始,减轻严重性或治愈疾病,提高生存率或更快的恢复,或防止疾病发生,或者上述中的两种或更多种的组合。
[0077]
如本文使用的术语“有效量”由诸如本领域中可能已知的考虑因素来确定。该量必须是有效的以达到如上文描述的期望的治疗效果,这尤其取决于待治疗疾病的类型和严重性以及治疗方案。有效量通常在适当设计的临床试验(剂量范围研究)中被确定,并且本领域技术人员将知道如何恰当地进行这样的试验以便确定有效量。如通常已知的,有效量取决于多种因素,包括配体对受体的亲和力、其在体内的分布概况、多种药理学参数诸如在体内的半衰期、不期望的副作用(如果有的话)、诸如年龄和性别的因素,等等。
[0078]
抗生素剂可以以一定的量或剂量存在于本发明的制剂中,所述量将取决于药物制剂领域的技术人员已知的多种考虑因素。不希望受任何剂量量的限制,通常抗生素剂可以以在0.1%w/w和75%w/w之间的量存在,这取决于药物的效能、配置用于例如注射施用或局部施用的制剂的体积以及期望的释放概况。本发明的聚合物的疏水特性可以部分地保护并入的药物在储存期间和在患者中免受由于光相互作用、氧化或水解引起的劣化。糊状聚合物可以采用本领域中已知的施用方法被注射或散布在患病的表面上,诸如肺、结肠和其他组织。
[0079]
本发明的制剂可以通过多种方式递送。在一些实施方案中,有效量的抗生素剂可以局部、口服或通过注射来施用。在一些实施方案中,施用是通过以下途径中的一种或更多种:口服、局部、经粘膜、经鼻、肠内、胃肠外、肌肉内、皮下、髓内注射、鞘内、直接心室内、静脉内、腹膜内、鼻内或眼内注射。
[0080]
在一些实施方案中,制剂通过注射施用。
[0081]
在一些实施方案中,制剂可以经由使用以下来施用:片剂、丸剂、胶囊、小球、颗粒剂、粉末、锭剂、小袋、扁囊剂、酏剂、混悬剂、分散剂、乳剂、溶液、糖浆、气雾剂、凝胶、软膏、洗剂、乳膏和栓剂。
[0082]
为了实现全身施用,制剂可以经由口服、直肠、经皮、肠胃外(皮下、腹膜内、静脉内、动脉内、经皮和肌肉内)、局部、鼻内或经由栓剂施用来施用。
[0083]
在一些实施方案中,施用是对患病的组织或器官的部位或者靠近或邻近该部位的局部施用。局部施用可以是局部地或通过注射。
[0084]
如本文使用的、关于注射或递送的部位或者局部施用的部位的术语“局部的”以及术语“靠近”或“邻近”,指的是距患病的组织或器官的部位约0cm至约10cm的半径。
[0085]
根据本发明的使用方法和用途利用了呈式-(sa-ra)n-的聚酸酐的形式的本发明
的载体,其中sa是癸二酸并且ra是蓖麻油酸,并且其中n是在10和100之间的整数,所述聚酸酐具有低于2.5或低于2的mw/mn值(其中mw是重均分子量并且mn是数均分子量)或在1和2.5之间或1和2之间的值。
[0086]
在一些实施方案中,载体通过本文公开的任何工艺来制备。
[0087]
在一些实施方案中,根据本发明使用的制剂包含如所定义的抗生素剂和如所定义的载体,其中所述载体通过包括以下的工艺来制备:在存在每个游离羧酸基团不超过其摩尔当量的量的乙酸酐的情况下和在不存在溶剂的情况下ra和sa的熔融缩聚。
[0088]
附图简述
[0089]
参照以下附图,在阅读本发明的非限制性示例性实施方案的以下详细描述之后,可以更清楚地理解本发明,在附图中:
[0090]
图1是本发明的聚酸酐载体的合成方案。
[0091]
发明详述
[0092]
实施例1:形成根据本发明的载体的不同类型的二羧酸和羟基酸的低聚物的受控合成
[0093]
目标:合成不同类型的二羧酸和羟基酸的低聚物的可选择的方法的开发。
[0094]
材料:辛二酸(sua)和十二烷二酸(ddda)按原样收到的使用。由蓖麻油的水解制备蓖麻油酸(ra),如合成部分中描述的。
[0095]
光谱分析在varian 300mhz nmr光谱仪上使用cdcl3作为溶剂获得1h nmr谱和
13
c nmr谱,所述溶剂包含四甲基硅烷作为位移参照。使用具有金刚石晶体的nicolet is10光谱仪(thermo scientific,massachusetts)的智能itr atr取样附件进行傅立叶变换红外(ftir)光谱法。
[0096]
从蓖麻油制备蓖麻油酸:在1000ml圆底烧瓶中,通过加热(65℃)将48g的koh溶解在400ml的乙醇中。然后,向其中加入200g的蓖麻油,并且将它们适当地混合。然后将混合物在连续搅拌的情况下在140℃回流持续2h。在回流之后,通过蒸发器蒸发溶剂。然后加入200ml的双蒸水、150ml二异丙醚和150ml h3po4,并且将全部混合物转移到分液漏斗中。然后将其用双蒸水反复洗涤(3次-5次,每次200ml),直到水相的ph~4。然后,有机相通过磷酸钠收集并且蒸发至干燥,以获得纯的185g的蓖麻油酸(收率92.5%),其通过1h nmr证实。
[0097]
sua-ra和ddda-ra低聚物的合成:通过辛二酸和十二烷二酸与蓖麻油酸在170℃的酯化反应来合成sua-ra和ddda-ra低聚物。在圆底烧瓶中,取15g的sua、15g的ra和催化量(1%)的磷酸,并且在氮气下加热至170℃持续5小时。然后将另外15g的ra加入到圆底烧瓶中,并且在氮气急流(nitrogen swift)下继续加热持续另外的4小时。最后,加入另外5g的ra,并且再次在真空下在混合的情况下继续加热过夜,得到具有30:70的sua和ra比的sua-ra低聚物,其通过1h nmr表征。按照相同的程序合成了具有30:70的ddda和ra比的ddda-ra低聚物,并且也通过1h nmr表征。
[0098]
结果的讨论使用两种不同的二羧酸和羟基酸合成了两种不同的低聚物。在熔融和真空条件下,用sua或ddda酯化ra,其中h3po4被用作催化剂。在该反应条件下,100%的ra在与sua或ddda的酯化反应中被消耗,这由1h nmr证实,因为在最后的酯化步骤之后,醇质子在3.6ppm处的信号失去。此外,还避免了该方案中ra的自缩合(经由将ra逐步添加到sua或ddda中);证据来自1h nmr,因为在4.1ppm处没有信号。因此,该工艺给出了明确的sua-ra或
ddda-ra低聚物,没有任何残余物或自缩合的ra。
[0099]
实施例2:从可选择的方法研究聚(酯-酸酐)的合成
[0100]
目的是开发合成可生物降解的聚(酯-酸酐)的共聚物的可选择的方法。这里的重点在于三个特征:
[0101]
1)使用癸二酸(sa)和蓖麻油酸(ra)或12-羟基硬脂酸(hsa)以通过直接缩合来制备sa-ra或sa-hsa低聚物。
[0102]
2)使用较少量(1:1当量或更少)的乙酸酐来活化低聚物用于聚合。
[0103]
3)根据用于预聚合步骤的乙酸酐的量来控制聚(酯-酸酐)的分子量。
[0104]
材料:癸二酸(sa,99%纯度;aldrich,usa)、12-羟基硬脂酸(hsa)和乙酸酐(merck,德国)按原样收到的使用。由蓖麻油的水解制备蓖麻油酸(ra),如合成部分中描述的。
[0105]
光谱分析:在varian 300mhz nmr光谱仪上使用cdcl3作为溶剂获得1h nmr谱和
13
c nmr谱,所述溶剂包含四甲基硅烷作为位移参照。使用具有金刚石晶体的nicolet is10光谱仪(thermo scientific,massachusetts)的智能itr atr取样附件进行傅立叶变换红外(ftir)光谱法。
[0106]
分子量测定:通过凝胶渗透色谱法(gpc)系统waters 1515测定分子量。等度hplc泵,具有waters 2410折射率检测器、waters 717plus自动进样器和带有20μl回路的rheodyne(cotati,ca)注射阀。用chcl3(hplc级)通过线性styragel hr5柱(waters)以1ml/min的流量来洗脱样品。相对于聚苯乙烯标准品测定分子量。
[0107]
合成和表征:sa-ra低聚物:通过在175℃加热蓖麻油酸和癸二酸来合成sa-ra低聚物。在圆底烧瓶中,取30g的sa、30g的ra和催化量(0.1%)的磷酸,并且在氮气下加热至170℃持续5小时。然后将另外30g的ra加入到圆底烧瓶中,并且在氮气急流下继续加热持续另外的4小时。最后,加入另外10g的ra,并且再次在真空下在混合的情况下继续加热过夜,得到具有30:70的sa和ra比的sa-ra低聚物,其通过1h nmr和ftir表征。还通过相同的工艺制备了不同比的sa-ra低聚物,并且通过1h nmr表征。细节在下表1中给出。
[0108]
表1:sa-ra低聚物
[0109][0110]
sa-hsa低聚物
[0111]
还通过在175℃加热12-羟基硬脂酸和癸二酸来合成sa-hsa低聚物。在圆底烧瓶中,取15g的sa、15g的hsa和催化量(0.1%)的磷酸,并且在氮气下加热至170℃持续5小时。然后将另外15g的hsa加入到圆底烧瓶中,并且在氮气急流下继续加热持续另外的4小时。最后,加入另外5g的hsa,并且再次在真空下在混合的情况下继续加热过夜,得到具有30:70的sa和hsa比的sa-hsa低聚物,其通过1h nmr和ftir表征。还通过相同的工艺制备了20:80比的sa-hsa低聚物。细节在下表2中给出。
[0112]
表2:sa-ra低聚物
[0113][0114]
聚(sa-ra)
[0115]
在典型的合成中,10g的20:80、25:75、30:70、35:65比的sa-ra低聚物在140℃在氮气气氛下单独地熔融。然后向熔化的sa-ra低聚物中加入1:5当量的乙酸酐,并且在140℃回流持续60min。蒸发过量的乙酸酐或乙酸。然后使残余物经历在160℃在10毫巴下的熔融缩合持续4小时。还在相同的程序下使30:70比的sa-ra低聚物聚合,其中使用不同量(1当量、0.7当量、0.5当量、0.35当量、0.25当量、0.15当量)的乙酸酐(在140℃回流,过夜),以使用较少量的乙酸酐并且控制分子量。
[0116]
聚(sa-hsa)
[0117]
按照与聚(sa-ra)相同的程序,10g的20:80和30:70比的sa-hsa低聚物在140℃在氮气气氛下单独地熔融。然后向两种熔化的sa-hsa低聚物中加入1:5当量的乙酸酐,并且在140℃回流持续60min。蒸发过量的乙酸酐或乙酸。然后使残余物经历在160℃在真空(~10毫巴)下的熔融缩合持续4h。
[0118]
结果的讨论
[0119]
通过无溶剂的熔融缩聚工艺合成了两种聚(酯-酸酐)共聚物,其中直接使用癸二酸代替使用聚(sa)作为起始材料来合成sa-ra或sa-hsa低聚物。在熔融和真空条件下用sa酯化ra或has,其中使用约1%h3po4作为催化剂。在该反应条件下,100%的ra或hsa在与sa的酯化反应中被消耗,这由1h nmr证实,因为在最后的酯化步骤之后,醇质子在3.6ppm处的信号失去。此外,还避免了该方案中ra或hsa的自缩合(经由将ra或hsa逐步添加到sa中);证据来自1h nmr,因为在4.1ppm处没有信号。因此,该工艺给出了明确的sa-ra或sa-hsa低聚物,没有任何残余物或自缩合的ra或hsa。在~4.8ppm观察到酯化的聚合物的质子化学位移。与酯键和酸酐键相邻的两个质子分别出现在2.43ppm和2.33ppm。
[0120]
通过gpc测定了如所合成的聚合物(as-synthesized polymer)的分子量。分子量和不均衡性(disparity)的细节在下表3中给出,并且对分子量的控制取决于所用的乙酸酐。
[0121]
表3:本发明的聚合物的分子量和不均衡性
[0122]
[0123][0124]
实施例3:具有减少的反应时间的聚(sa-ra)的合成
[0125]
目标:该方案的目标是经由1h nmr监测聚(癸二酸-蓖麻油酸)的可生物降解的共聚物的合成过程,以减少反应时间。
[0126]
材料:癸二酸(sa,99%纯度;aldrich,usa)按原样收到的使用。由蓖麻油的水解制备蓖麻油酸(ra),如合成部分中描述的。
[0127]
光谱分析:使用cdcl3作为溶剂,在varian 300mhz nmr光谱仪上获得1h nmr谱。使用具有金刚石晶体的nicolet is10光谱仪(thermo scientific,massachusetts)的智能itr atr取样附件进行傅立叶变换红外(ftir)光谱法。
[0128]
分子量测定:通过凝胶渗透色谱法(gpc)系统waters 1515测定分子量。等度hplc泵,具有waters 2410折射率检测器、waters 717plus自动进样器和带有20μl回路的rheodyne(cotati,ca)注射阀。用chcl3(hplc级)通过线性styragel hr5柱(waters)以1ml/min的流量来洗脱样品。相对于聚苯乙烯标准品测定分子量。
[0129]
sa-ra低聚物的合成:通过在170℃加热蓖麻油酸和癸二酸来合成sa-ra低聚物。在圆底烧瓶中,取15g的sa、15g的ra和催化量(0.1%)的磷酸,并且在氮气下加热至170℃持续2小时。然后将另外15g的ra加入到圆底烧瓶中,并且在真空下继续加热持续另外的2小时,随后是氮气急流持续15min。最后,加入5g的ra,并且再次在真空下继续加热持续另外8小时,得到具有30:70w/w的sa和ra比的sa-ra低聚物,其通过1h nmr表征。
[0130]
聚(sa-ra):在典型的合成中,10g具有30:70比的sa-ra低聚物在氮气气氛下在140℃熔融。然后,将相对于低聚物中的酸1当量的乙酸酐加入到熔化的sa-ra低聚物中,并且在140℃回流持续2小时。蒸发过量的乙酸酐或乙酸。然后使残余物经历在160℃在真空(~10毫巴)下的熔融缩合持续4小时。
[0131]
结果的讨论:
[0132]
在熔融和真空条件下,用sa酯化ra,其中h3po4被用作催化剂。在此反应条件下,在与sa的酯化反应中,在12小时内100%的ra被消耗。这通过1h nmr证实,因为在最后的酯化步骤之后,醇质子在3.6ppm处的信号失去。此外,还避免了该方案中ra的自缩合(经由将ra逐步添加到sa中);证据来自1h nmr,因为在4.1ppm处没有信号。然后通过在140℃与1当量的乙酸酐回流持续2小时,随后在160℃在真空下加热持续4小时,使低聚物聚合。通过gpc测量聚合物的分子量,并且与以下的聚合物进行比较:所述聚合物由相同的具有30:70比的sa-ra低聚物通过在140℃与1当量的乙酸酐回流过夜、随后在160℃在真空下加热持续4小时来合成。注意到这两种工艺给出几乎相同的聚合物分子量(~11500道尔顿)。
[0133]
实施例4:庆大霉素从不同聚合物批次中的体外释放
[0134]
本研究的目的是确定由通过本发明的方法制备的、其中ra:sa低聚物的乙酸酐与羧酸的摩尔比为0.8的聚(sa=ra)30:70糊状聚合物制备的和由所制备的其中该比为5的聚合物制备的庆大霉素制剂之间的差异。研究了庆大霉素从负载在不同批次的聚(sa=ra)30:70糊状聚合物中的20%硫酸庆大霉素在水性介质中的释放。该释放研究是在乙酸盐缓冲液ph4.5中确定的,该乙酸盐缓冲液被确定为对含氨基分子诸如庆大霉素的释放是有用的。为了比较,测定了在37℃在磷酸盐缓冲液ph7.4中的释放。
[0135]
使用五种聚合物样品,其使用0.80摩尔比的乙酸酐与羧酸和4小时的聚合时间、在160℃、在15mm hg真空下通过本发明的程序类似地制备,具有重均分子量mw=9400 /-300和1.35的多分散性。为了比较,在相同的聚合条件下,通过使用5摩尔比的乙酸酐与羧酸制备五种聚合物样品,其具有重均分子量mw=12000 /-4200和3.2的多分散性。本发明的聚合物的特性黏度为0.15 /-0.1,而通过旧程序制备的聚合物示出0.20 /-0.5的特性黏度。
[0136]
将这些聚合物用于如下制备20%负载的硫酸庆大霉素:庆大霉素(gm)首先通过在120℃加热持续1小时来干燥,并且然后允许在真空中冷却至室温。然后将该干燥的gm并入p(sa-ra)(30:70)中。该并入通过经由研磨将干燥的gm粉末(20%w/w)与聚合物混合直到形成均质的糊状物来进行。如果聚合物是黏性的,则应用将聚合物加热至40℃。将制剂负载在2ml玻璃注射器中,并且测定通过23g针头的可注射性。还将空白聚合物负载在注射器中用于可注射性测试。空白聚合物和由本发明制备的具有窄分子量和多分散性的庆大霉素负载的聚合物制剂示出相同的可注射性,其中在柱塞上施加相同的力的情况下,聚合物或制剂从注射器和针头中平稳释放。负载有聚合物的注射器和负载有旧方法的具有宽分子量和多分散性的制剂的注射器是不一致的,只有两个注射器能够使用平常的对柱塞的力释放聚合物或制剂,而三个注射器没有可注射性并且需要额外的力来允许制剂穿过针头,其中一个聚合物注射器和一个制剂注射器在室温不允许任何释放。
[0137]
通过将1克的制剂放入用塑料网覆盖(小瓶的盖)并且沉降在800ml玻璃容器的底部的塑料容器中来测定体外释放。在乙酸盐缓冲液介质(ph=4.5)(由nacl:3.41克/升、乙酸:3.33ml/升、乙酸钠:3.41g/升组成)中在37℃以10rpm摇动进行释放研究。为了比较,还在37℃在磷酸盐缓冲液ph7.4中测定了释放。
[0138]
通过uv测定在释放介质中的gm分析。在1μg/ml-16μg/ml的浓度范围内制备校准曲线,其中gm与200μl在丙酮中的0.1mg/ml荧光胺溶液反应,使用硼酸盐缓冲液ph-7将样品体积补足到2ml,在室温孵育持续15min,并且在激发波长390nm和发射波长460nm处通过光谱荧光计进行分析。基于在从释放溶液中测定庆大霉素的同一天准备的校准曲线来计算释放的量。
[0139]
通过在制剂中加入20ml的氯仿来测定剩余制剂样品中的庆大霉素含量,并且涡旋持续两分钟。将混合物保持在37℃持续4小时,并且然后加入20ml的酸性ddw(ph=2),并且然后在涡旋下进行混合。为了得到两个分离的相,使用以4000rpm的离心持续10分钟。取样品的上部(ddw)相,以便分析在释放之后聚合物中剩余的gm的量。庆大霉素浓度通过使用荧光的分光荧光计来测定。可以从聚合物制剂中回收约80%的庆大霉素含量的回收率。
[0140]
庆大霉素在ph 4.5介质中不断地释放持续28天。每周用新的缓冲溶液替换释放介质溶液。在28天之后,测定剩余制剂中的庆大霉素含量。用本发明的聚合物制备的制剂在整
个28天内示出几乎线性的释放概况,每个数据点具有在1%和5%之间的窄标准偏差。约60%的gm被释放。从剩余的聚合物制剂中的庆大霉素的回收率是原始庆大霉素含量的约20%,没有获得完全回收。在中性ph磷酸盐缓冲液ph7.4中的释放在第一周是恒定的,其中约20%的gm被释放,并且随后由于在gm和不太水溶性的酸性降解产物低聚物之间可能的盐形成,仅很少的gm被释放。
[0141]
庆大霉素从旧方法的聚合物制剂中的释放在28天内是恒定的,但是标准偏差在每个时间点处释放的量的5%和20%之间。庆大霉素从这些聚合物制剂中的回收率在原始含量的10%和25%之间。
[0142]
实施例5:庆大霉素从辐照这的制剂和未辐照过的制剂中的释放速率的比较。
[0143]
在本研究中使用了在p(sa:ra)(30:70)中具有20%(w/w)的负载的庆大霉素制剂和具有2.5mrad剂量的负载在玻璃注射器中的在辐照之后的制剂。将200mg制剂负载在塑料盖中,该塑料盖具有约1.76cm2的覆盖面积并且浸入100ml磷酸盐缓冲盐水ph 7.4中,该磷酸盐缓冲盐水由nacl 8g/l、kcl 0.2g/l、na2hpo
4 12h2o 2.9g/l、kh2po
4 0.24g/l组成。将小瓶放在37℃的烘箱中的定轨振荡器(orbital shaker)30rpm上。在1小时、8小时、24小时、48小时、72小时和168小时之后取2ml的样品。在24小时、7小时2、168小时之后,由新的缓冲液替换介质。
[0144]
辐照过的样品和未辐照过的样品两者具有相似的黏度,并且分子量或外观没有变化。在研究期间,两者都以恒定的方式释放负载的gm。约50%的gm被释放,其中约15%-20%的药物从剩余的制剂中回收。在释放研究之前制剂的ft-ir光谱指示存在酯键和酸酐键,并且在168小时的释放之后,仅很少的酸酐键在聚合物中,但有高的酯峰和羧酸峰。辐照过的制剂和未辐照过的制剂两者示出相似的ftir光谱。
[0145]
实施例6:硫酸庆大霉素负载的psa:ra 30:70的毒性
[0146]
潜在毒性测试项目:在本发明的psa:ra 30:70糊状物和空白聚合物载体中10%和20%负载的硫酸庆大霉素。将这些制剂皮下注射至sprague dawley大鼠以测定mtd。
[0147]
研究如下进行:6组大鼠,每组中6只大鼠,3只雄性和3只雌性。三组注射有0.2ml的空白聚合物、10%负载的庆大霉素和20%庆大霉素。其余三组是相同的,但注射剂量是0.4ml。动物被追踪持续14天并且处死。在研究结束时,进行一般尸体解剖,并且提交注射部位的皮肤用于组织病理学。
[0148]
提供给药材料以备用。每种给药材料在给药的当天解冻,并且经由柱塞端直接从注射器转移到预装有19g薄壁针头的注射器中。在整个14天的研究期间,任何经治疗的动物或安慰剂和盐水对照的动物都没发生死亡。
[0149]
在任何测试项目中均未观察到明显的处理相关的全身反应。从给药当天起并且直到给药后14天的预先安排的处死,在分配到研究的所有动物中观察到注射部位处呈皮下肿块(subcutaneous bulge)的形式的局部反应。在整个14天的观察期内,在任何盐水对照动物中均未观察到局部反应。
[0150]
所有动物在14天的时间段结束时均发生预期的体重变化。
[0151]
在尸体解剖时,所有测试的动物均展示出囊状团块(capsule-like mass),其通常填充有坚硬物质。盐水对照处理的动物未示出严重的病理学发现。
[0152]
组织病理学评估揭示出,所有测试组中在注射部位处在大小和特性方面相当的组
织反应。反应包括中央腔区域,其由肉芽肿性炎症的层围绕,并且更外部是纤维化层。建议空腔(大部分为3级-中等)来反映冲掉的注射的材料。肉芽肿性炎症层(大部分为2级-轻度)包括单核细胞、巨噬细胞和多核巨细胞的混合物。纤维化层(大部分为3级-中度)包括包埋在胶原中的成纤维细胞。预期肉芽肿性反应在逐渐被吸收的异物的注射之后被看到。对于庆大霉素植入系统,与注射有安慰剂的组相比,庆大霉素的存在与炎症的特性、级别和范围的任何增加无关。在注射有盐水的动物组中未观察到损伤。
[0153]
实施例7:包含20%w/w庆大霉素的p(ra-sa)的效力评价
[0154]
测试了包含20%w/w庆大霉素的p(ra-sa)消除细菌和减少骨髓炎对骨愈合的负面影响的有效性。从包含20%w/w庆大霉素的p(ra-sa)中释放的抗生素,庆大霉素,是氨基糖苷类,由于热稳定性和宽的抗菌谱,其通常用于人类模型和动物模型两者中以治疗或预防骨髓炎。建立了用于金黄色葡萄球菌人为污染的(artificial contaminated)开放性骨折的截骨模型(osteotomy model)。与创伤性骨折的诱导相比,该模型在多种动物中提供了改进的再现性。由于低的机械负担,使用了桡骨。此外,周围的肌肉和平行的尺骨有助于骨折的机械支撑,无需另外的固定器。有效治疗的标准是在从接种和治疗的部位分离的骨的骨悬浮液(bone suspension)中的金黄色葡萄球菌计数。使用三组动物,每组中6只,一组不治疗,一组仅用聚合物治疗,并且一组用包含20%w/w庆大霉素的p(ra-sa)治疗。实验持续28天,其中在最后一天,处死动物并且将在金黄色葡萄球菌接种的部位处的骨分离,粉碎到在灭菌的缓冲溶液中的悬浮液中,并且测试细菌含量。
[0155]
在第1组中的所有6个样品(被污染的,没有治疗)均是金黄色葡萄球菌阳性的并且与第2组中的样品(仅p(ra-sa))相似地繁殖。检测到》1000cfu/ml的金黄色葡萄球菌计数。在第3组(被污染的,用包含20%w/w庆大霉素的p(ra-sa)局部治疗)中,所有6只动物均是金黄色葡萄球菌阴性的。因此,采用包含20%w/w庆大霉素的p(ra-sa)的局部治疗成功地完全根除了用来诱导污染的金黄色葡萄球菌细菌。因此,微生物检查揭示出,施用包含20%w/w庆大霉素的p(ra-sa)导致优良的培养评价,其中在任何样品中均没有金黄色葡萄球菌的生长。
[0156]
在整个28天的观察期内,没有发生任何动物的死亡。在骨折诱导后的前几天期间,观察到回避使用被操作过的肢体(operated limb)。在第三周和第四周期间,在来自第3组的1只动物以及第1组和第2组的所有动物中观察到这种行为。在第1对照组和第2对照组的大多数动物中看到治疗过的骨的肿胀。在骨折诱导后的第一周期间,注意到所有动物的体重的下降,然而,到第9天,所有动物恢复了其初始体重,并且表现出预期的生长模式,直到观察期的结束。
[0157]
实施例8:抗微生物剂的体外释放
[0158]
将以下抗微生物剂并入本发明的p(ra-sa)70:30中:妥布霉素、红霉素、万古霉素、环丙沙星、氯己定、两性霉素b、头孢呋辛、酮康唑、左氧氟沙星、克林霉素、阿奇霉素和阿昔洛韦。所有的剂都是干燥粉末,其在室温以5%w/w、10%w/w和20%w/w的浓度在聚合物糊状物中被手动混合,并且被负载在2ml玻璃注射器中,并且测定了穿过19g针头的可注射性、释放概况和在室温经过三个月的稳定性。对于所有的剂,获得具有不同黏度的均匀不透明糊状物。当药物含量从5%增加到20%时,注意到所得到的糊状物的黏度的增加。所有制剂均示出良好的可注射性,并且在3个月的储存期间,没有示出外观、药物含量和黏度的任何变
化。评估体外释放持续一周,其中将释放介质调节为释放的剂,并且在释放介质中的浓度主要通过uv来测定。所有的剂均示出持续释放,其中在释放研究期间,5%至50%的负载的剂被释放。通常,药物含量越高,观察到的释放越快。两性霉素b,一种高度水不溶性的剂,仅释放极少量到磷酸盐缓冲溶液中,然而,当向释放介质中加入1%span 80时,获得了较快的释放。
[0159]
实施例9:体内庆大霉素释放和聚合物消
[0160]
本研究的目的是测定庆大霉素向注射部位和血液中的释放,以及聚合物从注射部位的消除。给药后对动物进行临床观察持续多达8周。在各个观察期结束时和在其他预定时间点,从每只动物收集血浆、注射部位和周围区域,并且转移用于庆大霉素分析和组织病理学。
[0161]
在对雄性nzw兔进行单次肌肉内注射包含20%w/w庆大霉素的p(ra-sa)之后的多个时间点,注射部位的血液样品和肌肉样品用于评估庆大霉素从载体聚合物中局部释放的程度。
[0162]
使用每个注射器包含0.5ml制剂的预填充的注射器。制剂以0.2ml/只动物通过肌肉内途径被施用于麻醉的动物。制剂通过向右侧中部椎旁肌肉(right mid paravertebral muscle)(距脊髓~2.5cm-5cm,并且深度约1cm)的单次缓慢注射被施用。所有动物保持其体重,或者在没有临床疾病体征的情况下增加体重。所有血浆样品和肌肉样品均按计划收集。
[0163]
庆大霉素仅在注射后的前24小时期间在血液中以非常低的水平被发现,并且此后低于检测水平。在注射点的4mm直径的注射部位处的肌肉中的庆大霉素浓度在前3周内示出》100微克每克组织的非常高的浓度,并且在之后的几周内降低到~5-10微克每克组织。在距注射部位10mm和15mm的距离处的组织中,庆大霉素浓度显著降低。在8周结束时,注射部位的组织病理学指示仅有较少的炎症迹象,其中在注射部位处仅有微量的聚合物制剂。
[0164]
在大鼠中进行了类似的研究,其中用0.05ml的聚合物-庆大霉素制剂对大鼠进行肌肉内注射,并且测定在注射部位的肌肉处的庆大霉素血液水平。在前24小时期间,在注射部位处发现》1000微克每克组织的浓度。在注射部位处的药物浓度随时间呈指数下降,其中在3周之后浓度是~100微克每克组织,并且在8周之后~5微克每克组织。在注射后2小时发现1-8微克每ml的庆大霉素血液水平,并且在6小时之后低于检测水平。在研究期间,始终未观察到毒性的迹象。在8周之后注射部位的组织病理学确实示出几乎完全的愈合,其中几乎没有注射的材料的迹象。这些研究表明庆大霉素在注射部位的受控释放,其中在注射后6小时之后没有全身分布。
[0165]
实施例10:在用不同量的乙酸酐制备的聚酸酐中的乙酸/乙酸酐含量
[0166]
通过gcms测定了用过量的1:5乙酸酐与低聚物羧酸含量或用0.8:1的摩尔比合成的聚(ra:sa)70:30中的乙酸酐浓度和乙酸含量。将聚合物溶解在二氯甲烷中,并且立即注射到gcms中用于乙酸/乙酸酐测定。用过量的乙酸酐制备的聚合物示出在10ppm-100ppm的范围内的微量的乙酸/乙酸酐,而用摩尔当量或更少的乙酸酐制备的聚合物在聚合物样品中没有示出任何乙酸/乙酸酐。包含乙酸或乙酸酐的聚合物可能与并入的药物反应以形成新的分子实体或者被释放并且降低周围组织中的ph,这可能使组织感染。
[0167]
实施例11:聚(ra:sa)70:30的储存期稳定性
[0168]
将包装在铝箔封套中的在真空下的负载有0.5g聚(ra:sa)70:30糊状物的玻璃注
射器放在以下温度的柜中:-20℃、4℃-8℃和25℃,并且通过gpc测定分子量,通过黏度计测定黏度,并且通过ftir测定酸酐键含量。没有观察到分子量、黏度和ftir光谱的变化。
[0169]
本发明的聚合物在室温持续数月是稳定的,具有窄多分散性的批次间的高再现性,不包含微量的乙酸或乙酸酐,活性剂的并入是在室温在温和混合的情况下,多种粉状的剂可以配制在聚合物中并且获得的糊状制剂是可注射的,20%或甚至30%的药物负载是可能的,具有并入的剂的高度可再现的批次间释放概况,体外降解概况的高再现性。本发明的聚合物是高度生物相容性的,降解成容易从体内消除的天然脂肪酸。聚合物载体将并入的药物的释放限制在注射部位,并入的剂具有极少的全身分布。此外,两种或更多种活性剂可以并入聚合物并且从聚合物中释放,用于受控释放应用。本发明的聚合物不受辐照灭菌影响。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。