1.本发明涉及包含倍癌霉素(duocarmycin)衍生物的抗体-药物缀合物(conjugate)和硫代硫酸盐(thiosulfate)在治疗人肿瘤中的组合用途,由此硫代硫酸盐预防或降低所述抗体-药物缀合物的非期望的非靶组织毒性。
背景技术:
2.倍癌霉素是抗肿瘤抗生素家族的成员,包括倍癌霉素a、倍癌霉素sa和cc-1065。它们以其强大的抗肿瘤特性而闻名,但由于其极高的毒性,通常不被单独使用。目前,正在将倍癌霉素作为抗体-药物缀合物(antibody-drug conjugate,adc)中的细胞毒性药物进行探索。
3.adc有望通过将高效细胞毒性药物特异性地导向癌细胞来解决癌症有效新治疗中巨大的未满足的需求,从而提高疗效,同时降低小分子药物潜在的全身性毒副作用。
4.接头-药物vc-seco-duba
[0005][0006]
首次在wo2011/133039的p.210,ll.21-27中作为化合物18b公开。其是高效cc-1065类似物的一个实例。vc-seco-duba与抗her2抗体曲妥单抗(trastuzumab)的adc,即syd985或(vic-)曲妥珠单抗-多卡马嗪(trastuzumab duocarmazine),已被成功用于多项临床前研究(van der lee等人,molecular cancer therapeutics,2015,14(3),692-703;black等人,molecular cancer therapeutics,2016,15(8),1900-1909)和i期临床试验(clinicaltrials.gov nct02277717)。曲妥珠单抗-多卡马嗪目前正在tulip iii期临床试验中对her2阳性局部晚期或转移性乳腺癌患者进行测试(clinicaltrials.gov nct03262935)。
[0007]
与其他药物一样,曲妥珠单抗-多卡马嗪的使用也有非期望的非靶组织毒性。例如,在i期临床试验期间,观察到在静脉内输注过程中含有adc的溶液外渗所引起的眼部毒
性和局部毒性(banerji等人,lancet oncology,2019,20,1124-1135)。这种非期望的非靶组织毒性可以是非特异性的,即由以下情况引起:毒性药物在与靶标结合之前被过早释放,所释放的毒性药物从肿瘤细胞扩散到周围组织中,所谓的旁观者效应,或adc被非特异性地摄取到细胞中,例如巨胞饮作用(macropinocytosis)。或者,非靶组织毒性可以是抗原介导的:通过抗体(即曲妥珠单抗)与非肿瘤组织的细胞上表达的抗原靶标(即,her2)结合,随后adc被内化且细胞毒性药物在细胞内被释放。
[0008]
最近开始了另一种含有倍癌霉素衍生物的抗5t4 adc(即syd1875)的首次人体试验(clinicaltrials.gov nct04202705)。
[0009]
因此,通常来说需要预防和/或降低包含倍癌霉素衍生物的adc的非期望的非靶组织毒性,特别是包含vc-seco-duba的adc。
技术实现要素:
[0010]
本发明涉及包含倍癌霉素衍生物的adc和硫代硫酸盐在治疗人肿瘤中的组合用途,由此硫代硫酸盐预防或降低adc的非期望的非靶组织毒性。
[0011]
在第一方面,本发明涉及adc,其用于治疗人肿瘤,其中所述adc与硫代硫酸盐组合施用,并且其中所述adc是式(i)化合物
[0012][0013]
其中
[0014]
ab是抗体或抗体的抗原结合片段;
[0015]
n为0、1、2或3;
[0016]
m代表从1到6的平均药物-抗体比(drug-to-antibody ratio,dar);
[0017]
r1选自:
[0018]
[0019]
y是1至16的整数;并且
[0020]
r2选自:
[0021][0022]
在一个具体实施方案中,adc是式(ii)化合物
[0023][0024]
在第二方面,本发明涉及包含硫代硫酸盐的组合物,其用于在人中预防或降低与向人施用式(i)或(ii)的adc相关的毒性。
[0025]
在第三方面,本发明涉及式(i)或(ii)的adc在制备用于治疗人肿瘤的组合疗法的药物中的用途,其中所述药物与硫代硫酸盐组合施用。
[0026]
在第四方面,本发明涉及作为组合制剂的包含式(i)或(ii)的adc和硫代硫酸盐的产品,其用于同时、分开或依次治疗人肿瘤。
[0027]
在第五方面,本发明涉及预防或降低与施用式(i)或(ii)的adc相关的毒性的方法,其包括与有效量的硫代硫酸盐组合施用有效量的所述adc,其中所述硫代硫酸盐在所述adc首次施用之前约三周至之后约1小时施用,并且所述硫代硫酸盐的施用以规律间隔重复,直至所述adc最后一次施用之后最多三个月。
附图说明
[0028]
图1.seco化合物重排为含环丙基的化合物。
[0029]
图2.硫代硫酸盐对含环丙基化合物的解毒作用。
[0030]
图3a.syd986(1μm)空白溶液及syd986(1μm) 溶解在乙腈/水(1:1)中的sts(10mm)的uv色谱图。
[0031]
图3b.syd986空白(上图)和syd986与sts的反应产物(下图)的ms分析结果。
[0032]
图4a.暴露于多种浓度的syd986或多种浓度的syd986 1mm sts之后sk-br-3细胞的活力。
[0033]
图4b.暴露于多种浓度的syd1875或多种浓度的syd1875 1mm sts之后sk-br-3细胞的活力。
具体实施方式
[0034]
倍癌霉素是一类结构相关的毒素,最初是从链霉菌(streptomyces)物种的培养液中分离出来的。它们是抗肿瘤抗生素家族的成员,包括倍癌霉素a、倍癌霉素sa和cc-1065。倍癌霉素与dna的小沟结合,随后引起dna的不可逆烷基化。这破坏核酸结构,最终导致肿瘤细胞死亡。
[0035]
由于其极高的毒性,倍癌霉素及其合成衍生物通常不被单独使用,而是用作例如adc中的细胞毒性药物。
[0036]
曲妥珠单抗-多卡马嗪,一种adc,由式(iii)中抗体曲妥珠单抗和倍癌霉素衍生物接头-药物vc-seco-duba组成。
[0037][0038]
目前正在tulip iii期临床试验中进行测试(clinicaltrials.gov nct03262935)。关于曲妥珠单抗-多卡马嗪和其他包含倍癌霉素衍生物的adc(包括syd1875)开发的一个担忧是,细胞毒活性和随后的有效性可能与一些患者的实质性毒性相关。在i期临床研究中,例如,观察到了在静脉内输注期间adc外渗所引起的眼部毒性和局部毒性。曲妥珠单抗-多卡马嗪(syd985)和syd1875均是根据式(i)和(ii)的adc。
[0039]
本文中所用的术语“非期望的非靶组织毒性”意指对除靶组织之外的任何组织的毒性,即对非肿瘤组织的毒性,优选对健康细胞的毒性。非期望的非靶组织毒性可以是非特异性的或抗原介导的。
[0040]
非特异性的非期望的非靶组织毒性可以在以下情况发生,例如,通过旁观者效应或通过adc被非特异性摄取到细胞中,毒性药物从adc中被过早释放。
[0041]
从adc中过早释放毒性药物的一个实例是在循环中或在外渗后(即静脉内输注的adc渗漏到输注部位周围的血管外组织中),从adc中断裂出接头-药物。从adc中断裂出接头-药物导致活性毒素的形成,这可能会引发不良事件,例如组织坏死。旁观者效应是这样的事件,其中在已经结合和处理adc的细胞附近的没有结合和/或处理adc的细胞被杀伤。不希望受任何理论的束缚,认为药物是由垂死的细胞释放的,从而杀伤其附近的其他细胞。旁观者效应可以发生在影响adc所作用的肿瘤细胞附近的其他肿瘤细胞的肿瘤中,在此这是一种期望的效应。然而,它也可以发生在肿瘤内部或外部,由此杀伤位于adc所作用的(非)肿瘤细胞附近的非肿瘤细胞。在本发明中,术语旁观者效应仅限于其发生在肿瘤外部并影响非肿瘤细胞而不是在肿瘤附近的情况。非特异性摄取的一个实例是巨胞饮作用。巨胞饮作用是真核细胞摄取细胞外液体和溶解分子的一种手段。adc被细胞摄取后,细胞毒性药物在细胞内被释放,导致细胞死亡。
[0042]
非期望的非靶组织毒性也可以是抗原介导的,即在抗原也存在于除靶组织之外的其他组织上的情况下。不希望受任何理论束缚,认为adc中的抗体与在非肿瘤组织细胞上表达的抗原-靶标结合,adc随后被内化,接着细胞毒性药物在细胞内被释放且细胞死亡。
[0043]
惊人且出人意料的是,本发明人发现硫代硫酸盐可以对从adc释放的含活性环丙基的倍癌霉素药物(或过早释放的接头-药物)解毒,并且可以因此预防和/或降低非期望的非靶组织毒性。因此,本发明涉及包含倍癌霉素衍生物的adc和硫代硫酸盐的组合用途。
[0044]
因此,一方面,本发明涉及包含倍癌霉素衍生物的adc,其用于治疗人肿瘤,其中包含倍癌霉素衍生物的adc与硫代硫酸盐组合施用。适用于本发明的组合用途的adc包含如下定义的wo2010/062171中所公开的倍癌霉素衍生物。这样的adc在wo2010/062171和wo2011/133039中被一般性地公开并且可由下式描述
[0045]
ab-(l-d)m,
[0046]
其中ab是抗体或抗体的抗原结合片段,l-d是倍癌霉素衍生物接头-药物,并且m代表1至12的平均dar。
[0047]
wo2010/062171公开了一系列dna烷基化剂cc-1065的类似物。wo2010/062171的实施例1至22中描述了很多这些药物的化学合成。
[0048]
wo2010/062171中公开的倍癌霉素衍生物由dna结合(dna-binding,db)部分和式(iv)中描述的dna烷基化(dna-alkylating,da)部分组成
[0049][0050]
db部分选自
[0051][0052]
r2和r3独立地选自h、oh、sh、nh2、n3、no2、no、cf3、cn、c(o)nh2、c(o)h、c(o)oh、卤素、ra、sra、s(o)ra、s(o)2ra、s(o)ora、s(o)2ora、os(o)ra、os(o)2ra、os(o)ora、os(o)2ora、ora、nhra、n(ra)rb、
n(ra)(rb)rc、p(o)(ora)(orb)、op(o)(ora)(orb)、sirarbrc、c(o)ra、c(o)ora、c(o)n(ra)rb、oc(o)ra、oc(o)ora、oc(o)n(ra)rb、n(ra)c(o)rb、n(ra)c(o)orb、n(ra)c(o)n(rb)rc和水溶性基团,其中
[0053]
ra、rb和rc独立地选自h和任选地被取代的(ch2ch2o)
aa
ch2ch2x1r
a1
、c
1-15
烷基、c
1-15
杂烷基、c
3-15
环烷基、c
1-15
杂环烷基、c
5-15
芳基或c
1-15
杂芳基,其中aa选自1至1000,x1选自o、s和
nr
b1
,并且r
b1
和r
a1
独立地选自h和c
1-3
烷基、ra、rb和/或rc中的一个或更多个任选取代基任选地为水溶性基团,两个或更多个ra、rb和rc任选地通过一个或更多个键连接以形成一个或更多个任选地被取代的碳环和/或杂环。
[0054]
术语“水溶性基团”是指在水性环境中很好地溶剂化并且赋予其所连接的化合物改善的水溶性的官能团。水溶性基团的实例包括但不限于多元醇、直链或环状糖类、伯胺、仲胺、叔胺或季胺和多胺、硫酸根基团、磺酸根基团、亚磺酸根基团、羧酸根基团、磷酸根基团、膦酸根基团、次膦酸根基团、抗坏血酸根基团、乙二醇,包括聚乙二醇和聚醚。优选的水溶性基团是伯胺、仲胺、叔胺和季胺、羧酸根、膦酸根、磷酸根、磺酸根、硫酸根、-(ch2ch2o)
yy
ch2ch2x2r
yy
、-(ch2ch2o)
yy
ch2ch2x
2-、-x2(ch2ch2o)
yy
ch2ch
2-、乙二醇、低聚乙烯乙二醇和聚乙二醇,其中yy选自1至1000,x2选自o、s和nr
zz
,并且r
zz
和r
yy
独立地选自h和c
1-3
烷基。
[0055]
术语“被取代的”,当用作“烷基”、“杂烷基”、“环烷基”、“杂环烷基”、“芳基”、“杂芳基”等的形容词时,表示所述“烷基”、“杂烷基”、“环烷基”、“杂环烷基”、“芳基”、“杂芳基”或类似基团包含一个或多个取代基(通过取代氢引入)。示例性取代基包括但不限于oh、=o、=s、=nrd、=n-ord、sh、nh2、no2、no、n3、cf3、cn、ocn、scn、nco、ncs、c(o)nh2、c(o)h、c(o)oh、卤素、rd、srd、s(o)rd、s(o)ord、s(o)2rd、s(o)2ord、os(o)rd、os(o)ord、os(o)2rd、os(o)2ord、s(o)n(rd)re、os(o)n(rd)re、s(o)2n(rd)re、os(o)2n(rd)re、op(o)(ord)(ore)、p(o)(ord)(ore)、ord、nhrd、n(rd)re、
n(rd)(re)rf、si(rd)(re)(rf)、c(o)rd、c(o)ord、c(o)n(rd)re、oc(o)rd、oc(o)ord、oc(o)n(rd)re、n(rd)c(o)re、n(rd)c(o)ore、n(rd)c(o)n(re)rf、水溶性基团和这些取代基的硫代衍生物,以及任何这些取代基的质子化、带电和去质子化形式,其中rd、re和rf独立地选自h和任选地被取代的-(ch2ch2o)
yy
ch2ch2x2r
yy
、c
1-15
烷基、c
1-15
杂烷基、c
3-15
环烷基、c
1-15
杂环烷基、c
5-15
芳基或c
1-15
杂芳基或其组合,其中yy选自1至1000,x2独立地选自o、s和nr
zz
,并且r
zz
和r
yy
独立地选自h和c
1-3
烷基,rd、re和rf中的两个或更多个任选地通过一个或更多个键连接以形成一个或更多个任选地被取代的碳环和/或杂环。当存在多于一个取代基时,每个取代基都是独立选择的。两个或更多个取代基可以通过用一个或更多个连接键取代各个取代基上的一个或更多个氢原子而彼此连接,所述连接键可以是单键、双键或三键,或者,如果共振结构是可能的,则在两个或更多个这些共振结构中,所述键的键序(bond order)可以不同。因此,两个取代基可以在形成一个或更多个环的情况下连接。
[0056]
当取代基可以“通过一个或更多个键连接以形成一个或更多个任选地被取代的碳环和/或杂环”时,这意味着取代基可以通过一个或更多个连接键置换各个取代基上的一个或更多个氢原子。
[0057]
本文中所用的术语“芳基”是指包含5至24个环碳原子的碳环芳族取代基,其可以带电或不带电并且可以由一个环或两个或更多个稠合在一起的环组成。芳基的实例包括但不限于苯基、萘基和蒽基。
[0058]
本文中所用的术语“杂芳基”是指包含1至24个环碳原子和至少一个环杂原子例如氧、氮、硫、硅或磷的杂环芳族取代基,其中氮和硫可以任选地被氧化并且氮可以任选地被季铵化,所述杂芳基可以由一个环或两个或多个稠合在一起的环组成。杂原子可以直接相互连接。杂芳基的实例包括但不限于吡啶基、嘧啶基、呋喃基、吡咯基、三唑基、吡唑基、吡嗪基、噁唑基、异噁唑基、噻唑基、咪唑基、噻吩基、吲哚基、苯并呋喃基、苯并咪唑基、苯并噻唑基、嘌呤基、吲唑基、苯并三唑基,苯并异噁唑基、喹喔啉基、异喹啉基和喹啉基。在一个实施
方案中,杂芳基包含1至4个杂原子。应当指出的是,“c1杂芳基”是指杂芳基的环系中仅存在1个碳(因此不计入任选取代基中的碳原子)。这种杂芳基的一个实例是四唑基。
[0059]“芳基”和“杂芳基”基团还包括一个或更多个非芳族环和一个芳基或杂芳基环或环系统稠合的环系统。
[0060]
本文中所用的术语“烷基”是指直链或支链、饱和或不饱和的烃基取代基。烷基的实例包括但不限于甲基、乙基、丙基、丁基、戊基、己基、辛基、癸基、异丙基、仲丁基、异丁基、叔丁基、异戊基、2-甲基丁基、乙烯基、烯丙基、1-丁烯基、2-丁烯基、异丁烯基、1-戊烯基、2戊烯基和1-丁炔基。
[0061]
本文中所用的术语“杂烷基”是指直链或支链、饱和或不饱和的烃基取代基,其中至少一个碳原子被杂原子例如氧、氮、硫、硅或磷取代,其中氮和硫可以任选地被氧化,并且氮可以任选地被季铵化。杂原子可以直接相互连接。实例包括但不限于甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、叔丁氧基、甲氧基甲基、乙氧基甲基、甲氧基乙基、乙氧基乙基、甲氨基甲基、二甲氨基甲基、甲氨基乙基、二甲氨基乙基、甲硫基甲基、乙硫基甲基、乙硫基乙基和甲硫基乙基。
[0062]
本文中所用的术语“环烷基”是指饱和或不饱和的非芳族环状烃基取代基,其可以由一个环或两个或更多个稠合在一起的环组成。实例包括但不限于环丙基、环丁基、环戊基、环戊烯基、环戊二烯基、环己基、环己烯基、1,3-环己二烯基、十氢萘基和1,4-环己二烯基。
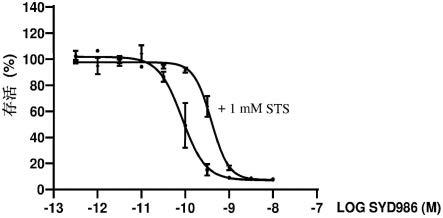
[0063]
本文中所用的术语“杂环烷基”是指饱和或不饱和的非芳族环状烃基取代基,其可以由一个环或两个或更多个稠合在一起的环组成,其中一个环中的至少一个碳被杂原子例如氧、氮、硫、硅或磷取代,其中氮和硫可以任选地被氧化,并且氮可以任选地被季铵化。杂原子可以直接相互连接。实例包括但不限于四氢呋喃基、吡咯烷基、哌啶基、1,4-二恶烷基、十氢喹啉基、哌嗪基、噁唑烷基和吗啉基。应当指出的是,“c1杂环烷基”表示杂环烷烃的环系中仅存在一个碳(因此不计入任选取代基中的碳原子)。这种基团的一个实例是二环氧乙烷基。
[0064]
本文中所用的术语“酰基”是指具有直链、支链或环状构型或其组合的基团,通过羰基官能团与母体结构连接。这种基团可以是饱和的或不饱和的、脂肪族的或芳香族的、碳环的或杂环的。c
1-c8酰基的实例包括乙酰基-、苯甲酰基-、烟酰基-、丙酰基-、异丁酰基-、草酰基-等。
[0065]“烷基”、“杂烷基”、“环烷基”、“杂环烷基”、“芳基”、“杂芳基”、“酰基”等可包含的碳原子数由所述术语之前的名称表示,即,c
1-10
烷基是指所述烷基可以包含1至10个碳(不计入连接到该烷基的任选取代基中的碳原子)。
[0066]
本文中的术语“碳环”是指饱和或不饱和的环烷烃或芳烃部分,其中术语“环烷烃”和“芳烃”分别定义为如上文所定义的“环烷基”和“芳基”取代基的母体部分。
[0067]
本文中的术语“杂环”是指饱和或不饱和的杂环烷烃或杂芳烃部分,其中术语“杂环烷烃”和“杂芳烃”分别定义为如上文所定义的“杂环烷基”和“杂芳基”取代基的母体部分。
[0068]
在例如与“烷基”相对的“亚烷基”中与
“‑
基”相对的延伸
“‑
亚基”表示所述例如“亚烷基”是通过至少一个或更多个双键或两个或更多个单键与一个或更多个其他部分连接的
二价(或多价)部分,而相对的所述例如“烷基”是通过一个单键连接到一个部分的单价基团。因此,术语“亚烷基”是指直链或支链的饱和或不饱和的亚烃基部分;本文中所用的术语“杂亚烷基”是指其中至少一个碳被杂原子取代的直链或支链、饱和或不饱和的亚烃基部分;本文中所用的术语“亚芳基”是指碳环芳族部分,其可由一个环或两个或更多个稠合在一起的环组成;本文中所用的术语“亚杂芳基”是指碳环芳族部分,其可以由一个环或两个或更多个稠合在一起的环组成,其中一个环中的至少一个碳被杂原子取代;本文中所用的术语“亚环烷基”是指饱和或不饱和的非芳族环状亚烃基部分,其可由一个环或两个或更多个稠合在一起的环组成;本文中所用的术语“杂环亚烷基”是指饱和或不饱和的非芳族环状亚烃基部分,其可以由一个环或两个或更多个稠合在一起的环组成,其中一个环中的至少一个碳被杂原子取代。示例性的二价部分包括上文针对其中一个氢原子被除去的一价基团给出的那些实例。
[0069]“聚亚烷基”、“聚亚烷基”、“聚亚芳基”、“聚亚杂芳基”、“聚亚环烷基”、“聚亚杂环烷基”等中的前缀“聚”表示两个或更多个这样的
“‑
亚基”部分,例如亚烷基部分连接在一起形成支链或非支链多价部分,其包含两个或更多个相邻部分的连接位点。类似地,例如低聚乙二醇中的前缀“低聚”表示两个或更多个乙二醇部分连接在一起形成支链或非支链多价部分。前缀“低聚”和“聚”的区别在于前缀“低聚”最常用于表示数量相对较少的重复单元,而前缀“聚”通常表示数量相对较多的重复单元。
[0070]
优选地,式(iv)的倍癌霉素衍生物是
[0071]
[0072][0073]
更优选地,r3选自h、甲基和甲氧基。甚至更优选地,r3是甲基。
[0074]
更优选地,式(iv)的倍癌霉素衍生物是
[0075][0076]
甚至更优选地,式(iv)的倍癌霉素衍生物是
[0077][0078]
在i期临床试验中观察到式(iii)的曲妥珠单抗多卡马嗪静脉施用外渗后引起的眼毒性和局部毒性后,本发明人旨在找到一种预防和/或降低这些毒性作用的方法,优选在观察到毒性的部位局部。
[0079]
据信式(iv)的倍癌霉素衍生物在体内被转化为活性的含环丙基化合物,同时伴随着hcl的消除,如图1所示(elgersma等人,molecular pharmaceutics,2015,12(6),1813-1835)。所得的含环丙基化合物是活性倍癌霉素衍生物药物,其发挥靶标特异性的治疗性毒性,以及非期望的非靶组织毒性。
[0080]
如实施例1所示,本发明示出硫代硫酸钠能够对曲妥珠单抗-多卡马嗪所释放的活性倍癌霉素衍生物药物(即syd986)解毒,而n-乙酰半胱氨酸、盐酸半胱胺、2-巯基乙磺酸钠盐(mercaptoethanesulfonic acid,mesna)和l-谷胱甘肽则不能。不希望受任何理论束缚,认为硫代硫酸钠能够通过与环丙基结构结合而对活性倍癌霉素衍生物药物解毒,如图2所示。
[0081]
式ab-(l-d)m的接头部分(-l-)可以是将药物(在本发明的上下文中指式(iv)的倍癌霉素衍生物)连接至抗体或抗原结合片段的任何已知或合适的部分。所述接头可以是线性的或非线性的,比如wo2018/069375中所公开的。这种接头可以是可断裂的或不可断裂的。通常,接头在某些条件下是可断裂的,以便如本领域已知的那样从抗体中释放药物,例如条件性可断裂或条件性可转变的部分,其可以通过化学、光化学、物理、生物或酶促过程而断裂或转变。
[0082]
为了能够将接头或接头-药物部分缀合至ab,(共价)结合至ab的接头末端通常包含一个可与ab的天然或非天然氨基酸在相对温和的条件下反应的官能团。该官能团在本文中称为反应性部分(reactive moiety,rm)。反应性部分的实例包括但不限于氨基甲酰卤、酰卤、活性酯、酸酐、α-卤代乙酰基、α-卤代乙酰胺、马来酰亚胺、异氰酸酯、异硫氰酸酯、二硫化物、硫醇、肼、酰肼、磺酰氯、醛、甲基酮、乙烯基砜、卤代甲基和甲基磺酸根。
[0083]
在本发明的一个优选实施方案中,rm是
[0084][0085]
其中
[0086]
x3选自-cl、-br、-i、-f、-oh、-o-n-琥珀酰亚胺、-o-(4-硝基苯基),
[0087]-o-五氟苯基、-o-四氟苯基、-o-c(o)-r4和-o-c(o)-or4;
[0088]
x4选自-cl、-br、-i、-o-甲磺酰基、-o-三甲磺酰基和-o-甲苯磺酰基;并且
[0089]
r4是支链或非支链的c
1-c
10
烷基或芳基。
[0090]
在一个优选的实施方案中,本发明涉及一种用于治疗哺乳动物,优选人的肿瘤的adc,其中所述adc与硫代硫酸盐组合施用,并且其中所述adc是式(i)化合物
[0091][0092]
其中
[0093]
ab是抗体或抗体的抗原结合片段;
[0094]
n为0、1、2或3;
[0095]
m代表从1到6的平均dar;
[0096]
r1选自:
[0097][0098]
y是1至16的整数;并且
[0099]
r2选自:
[0100][0101]
在一个优选实施方案中,n为0或1;
[0102]
m代表从1到4的平均dar;
[0103]
r1是
[0104][0105]
y是1至4的整数;并且
[0106]
r2选自:
[0107][0108]
在一个更优选的实施方案中,所述adc是式(ii)的化合物
[0109][0110]
在本发明的上下文中,式(i)和(ii)adc中的ab可以是任何抗体或其抗原结合片段,优选单克隆抗体(monoclonal antibody,mab)或其抗原结合片段。
[0111]
本文中所用的术语“抗体”优选指包含两条重链和两条轻链的抗体。通常,抗体或其任何抗原结合片段是指具有治疗活性的,但如adc领域已知的,这种独立的功效不是必需的。根据本发明使用的抗体可以是任何同型,例如iga、ige、igg或igm抗体。优选地,抗体是igg抗体,更优选地是igg1或igg2抗体。抗体可以是嵌合的、人源化的或人的。优选地,抗体是人源化的或人的。甚至更优选地,抗体是人源化或人igg抗体,更优选地是人源化或人igg
1 mab,最优选地是人源化igg
1 mab。抗体可具有κ(kappa)或λ(lambda)轻链,优选κ(kappa)轻链,即人源化或人igg
1-κ抗体。
[0112]
本文中所用的术语“抗原结合片段”包括fab、fab'、f(ab')2、fv、scfv或还原的igg(reduced igg,rigg)片段、单链(single chain,sc)抗体、单域(single domain,sd)抗体、双抗体或微型抗体(minibody)。
[0113]“人源化”形式的非人(例如啮齿动物)抗体是包含源自非人抗体的最少序列的抗体(例如非人-人嵌合抗体)。用于人源化非人抗体的多种方法是本领域已知的。例如,重链(heavy chain,hc)和轻链(light chain,lc)的可变区(variable region,vr)中的抗原结合互补决定区(complementarity determining regions,cdr)来源于非人物种(通常是小鼠、大鼠或兔)的抗体。这些非人cdrs可以与hc和lc可变区的人框架区(framework regions,fr,即fr1、fr2、fr3和fr4)组合,从而使抗体的功能特性,例如结合亲和力和特异性至少部分保留。人fr中的选定氨基酸可以与相应的原始非人物种氨基酸交换,以进一步改进抗体性能,例如提高结合亲和力,同时保持低免疫原性。如此人源化的可变区通常与人恒定区组合。用于非人抗体人源化的示例性方法是winter及其同事提出的方法(jones等人,nature 1986,321,522-525;riechmann等人,nature 1988,332,323-327;verhoeyen等人,science 1988,239,1534-1536)。或者,可以通过修饰非人抗体氨基酸序列以将其人源化,以提高与人天然产生的抗体变体的相似性。例如,将原始非人物种fr的选定氨基酸交换为其相应的人氨基酸以降低免疫原性,同时保留抗体的结合亲和力。更多细节参见jones等人,nature 1986,321,522-525;riechmann等人,nature 1988,332,323-327;和presta,curr.op.struct.biol.1992,2,593-596。另见以下综述文章和其中引用的参考文献:vaswani和hamilton,ann.allergy,asthma and immunol.1998,1,105-115;harris,biochem.soc.transactions 1995,23,1035-1038;和hurle和gross,curr.op.biotech.(1994),5,428-433。
[0114]
可以使用kabat的方法(kabat等人,sequences of proteins of immunological interest,第5版public health service,national institutes of health,bethesda,md,nih出版号91-3242,pp.662、680、689(1991)),chothia的方法(chothia等人,nature 1989,342,877-883)或imgt的方法(lefranc,the immunologist 1999,7,132-136)来确定cdr。
[0115]
通常,所述抗体是单特异性(即,对一种抗原具有特异性;这种抗原可以在物种间共有或在物种间具有相似的氨基酸序列)或双特异性(即,对一个物种的两种不同抗原具有特异性)抗体,其包含至少一个hc和lc可变区以与肿瘤相关抗原(tumor associated antigen,taa)结合。优选地,taa是膜结合的taa,其可以是内化的或不内化的,优选内化的。
[0116]
在一个具体实施方案中,taa选自:膜联蛋白a1、b7h3、b7h4、ca6、ca9、ca15-3、ca19-9、ca27-29、ca125、ca242(癌抗原242)、ccr2、ccr5、cd2、cd19、cd20、cd22、cd30(肿瘤
坏死因子8)、cd33、cd37、cd38(环状adp核糖水解酶)、cd40、cd44、cd47(整合素相关蛋白)、cd56(神经细胞黏附分子)、cd70、cd74、cd79、cd115(集落刺激因子1受体)、cd123(白介素-3受体)、cd138(多配体蛋白聚糖1)、cd203c(enpp3)、cd303、cd333、cdcp1、cea、ceacam、clca-1(c型凝集素样分子-1)、cll 1、c-met(肝细胞生长因子受体)、cripto、dll3、egfl、egfr、epcam、eph(例如epha2或ephb3)、etbr(b型内皮素受体)、fap、fcrl5(fc受体样蛋白5,cd307)、fgfr(例如,fgfr3)、folr1(叶酸受体α)、gcc(鸟苷酸环化酶c)、gpnmb、her2、p95her2、hmw-maa(高分子量黑色素瘤相关抗原)、整合素α(例如,αvβ3和αvβ5)、igf1r、tm4sf1(或l6)、lewis a样碳水化合物、lewis x、lewis y(cd174)、liv1、间皮素(msln)、mn(ca9)、muc1、muc16、napi2b、nectin-4、pd-1、pd-l1、psma、ptk7、slc44a4、steap-1、5t4(或tpbg,滋养层糖蛋白)、tf(组织因子、促凝血酶原激酶、cd142)、tf-ag、tag72、tnfr、trop2(肿瘤相关钙信号转导子2)、vegfr和vla。
[0117]
合适抗体的实例包括博纳吐单抗(blinatumomab)(cd19)、依帕珠单抗(epratuzumab)(cd22)、伊妥木单抗(iratumumab)和本妥昔单抗(brentuximab)(cd30)、伐达妥昔单抗(vadastuximab)(cd33)、tetulumab(cd37)、伊沙妥昔单抗(isatuximab)(cd38)、比伐珠单抗(bivatuzumab)(cd44)、洛沃妥珠单抗(lorvotuzumab)(cd56)、沃瑟妥珠单抗(vorsetuzumab)(cd70)、米拉珠单抗(milatuzumab)(cd74)、波妥珠单抗(polatuzumab)(cd79)、洛伐妥珠单抗(rovalpituzumab)(dll3)、伏妥昔单抗(futuximab)(egfr)、奥珀妥珠单抗(oportuzumab)(epcam)、法乐妥珠单抗(farletuzumab)(folr1)、格巴妥木单抗(glembatumumab)(gpnmb)、曲妥珠单抗和帕妥珠单抗(pertuzumab)(her2)、伊瑞西珠单抗(etaracizumab)(整联蛋白)、阿奈妥单抗(anetumab)(间皮素)、潘科曼单抗(pankomab)(muc1)、恩弗妥单抗(enfortumab)(连接蛋白-4)以及h8、a1、和a3(5t4抗原)。
[0118]
在一个更具体的实施方案中,本发明涉及一种如上文所述的adc化合物,其中包含在adc中的抗体是抗膜联蛋白a1抗体、抗b7h3抗体、抗cd115抗体、抗cd123抗体,抗cll-1抗体、抗c-met抗体、抗her2抗体、抗muc1抗体、抗psma抗体、抗5t4抗体或抗tf抗体,优选符合式(i)或(ii)的adc化合物。
[0119]
在一个优选的实施方案中,所述式(i)或(ii)化合物中的ab是抗her2抗体曲妥珠单抗。
[0120]
在一个更优选的实施方案中,所述adc是式(iii)的化合物
[0121][0122]
如果适用,抗体或其抗原结合片段可包含(1)经工程改造的恒定区,即可引入一个或更多个突变以例如延长半衰期,为接头-药物提供附着位点和/或提高或降低效应器功
能;或(2)经工程改造的可变区,即可以已经引入一个或多个突变以提供接头-药物的附着位点。抗体或其抗原结合片段可以通过重组、合成或通过其他已知的合适方法产生。
[0123]
用于本发明使用的adc可以是野生型的或位点特异性的,并且可以通过本领域已知的任何方法产生,如下例所示。
[0124]
野生型adc可通过将接头-药物通过例如抗体的赖氨酸ε-氨基与抗体或其抗原结合片段缀合来产生,优选使用包含胺反应性基团例如活化酯的接头-药物;活化酯与抗体或其抗原结合片段联结将产生adc。或者,可以使用本领域已知的方法和条件,通过链间二硫键还原来缀合半胱氨酸侧链的游离硫醇与接头-药物以产生野生型adc,参见例如,doronina等人,bioconjugate chem.2006,17,114-124。制造过程包括部分还原暴露在溶剂中的链间二硫化物,然后所得的硫醇用含迈克尔受体的接头-药物(如含马来酰亚胺的接头-药物、α-卤代乙酸酰胺或酯)修饰。半胱氨酸连接策略使得每个还原的二硫化物最多两个接头-药物。大多数人igg分子具有四个暴露于溶剂的二硫键,因此每个抗体是可能有整数范围为0到8个接头-药物的。每个抗体的接头-药物的确切数量是由二硫键还原的程度和随后的缀合反应中使用的接头-药物的摩尔当量数决定的。所有四个二硫键被完全还原后产生每个抗体具有八个接头-药物的均质构建体,而部分还原通常导致产生每个抗体具有零个、两个、四个、六或八个接头-药物的异质混合物。
[0125]
位点特异性adc优选通过在突变抗体或其抗原结合片段的合适位置上的工程化半胱氨酸残基的侧链上将接头-药物缀合至抗体或其抗原结合片段来产生。工程化的半胱氨酸通常被其他硫醇(例如半胱氨酸或谷胱甘肽)封端以形成二硫化物。这些封端的残基需要在与接头-药物发生连接之前解封(uncapped)。接头-药物与工程残基的连接可以通过以下方式实现:(1)通过还原天然链间和突变的二硫化物,然后使用温和的氧化剂(如cuso4或脱氢抗坏血酸)重新氧化天然链间半胱氨酸,然后将解封的工程化半胱氨酸与接头-药物标准缀合,或(2)通过使用温和的还原剂,其以比还原链间二硫键更高的速率还原突变体二硫键,然后将解封的工程化半胱氨酸与接头-药物进行标准缀合。在最佳条件下,每个抗体或其抗原结合片段与两个接头-药物(即药物与抗体的比率,drug-to-antibody ratio,dar为2)将被连接(如果一个半胱氨酸被工程化到mab或片段的hc或lc中)。用于位点特异性缀合接头-药物的合适方法可以是例如wo2015/177360描述的还原和再氧化过程、wo2017/137628描述的使用温和还原剂的方法和wo2018/215427描述的用于同时缀合还原的链间半胱氨酸和解封的工程化半胱氨酸的方法。
[0126]
在本发明的上下文中治疗的肿瘤优选是表达adc所靶向的抗原的肿瘤。这样的肿瘤可以是人实体瘤或血液恶性肿瘤。如上定义的adc可以治疗的肿瘤的实例可以包括但不限于乳腺癌、脑癌(例如,胶质母细胞瘤)、头颈癌、甲状腺癌、肾上腺癌(例如,神经母细胞瘤)、骨癌(如骨肉瘤)、眼癌、食道癌、胃癌、小肠癌、结直肠癌、尿路上皮癌(如膀胱癌或肾癌)、卵巢癌、子宫癌、阴道癌和宫颈癌、肺癌(尤其是非小细胞肺癌,non-small cell lung cancer,nsclc)和小细胞肺癌(small-cell lung cancer,sclc)、黑色素瘤、间皮瘤(尤其是恶性胸膜间皮瘤)、肝癌(如肝细胞癌)、胰腺癌、皮肤癌、睾丸癌、前列腺癌、急性髓性白血病(acute myeloid leukemia,aml)、慢性髓性白血病(chronic myeloid leukemia,cml)、慢性淋巴性白血病(chronic lymphatic leukemia,cll)、急性淋巴细胞白血病(acute lymphoblastic leukemia,all)、非霍奇金淋巴瘤(non-hodgkin’s lymphoma,nhl)(包括滤
泡性淋巴瘤(follicular lymphoma,fl)和弥漫性大b细胞淋巴瘤(diffuse large b-cell lymphoma,dlbcl))和多个骨髓瘤(multiple myeloma,mm)。
[0127]
在本发明的上下文中,“硫代硫酸盐”可以是任何形式的;即,它可以是硫代硫酸,例如硫代硫酸,或硫代硫酸盐,例如硫代硫酸铵、硫代硫酸钙、硫代硫酸钾或硫代硫酸钠(sodium thiosulfate,sts),每一种都含有阴离子部分s2o
32-和合适的反阳离子。优选地,硫代硫酸盐是硫代硫酸盐,更优选是sts。
[0128]
sts(也被称为次亚硫酸钠)是一种无机化合物,分子式为na2s2o3·
xh2o。通常,它的可用形式为五水合物na2s2o3·
5h2o。由于它被用作食品防腐剂,因此普通人群会广泛接触到这种被认为是无毒的化合物(mcgeer等人,journal of neurology and neuromedicine 2016,1,28-30)。sts在1930年代被用作氰化物中毒的解毒剂。另一些医疗用途包括治疗癣、花斑癣和其他皮肤真菌感染,以及降低顺铂的副作用,例如肾毒性和耳毒性,以及外渗引起的局部毒性。sts被列入世界卫生组织的基本药物清单,这是卫生系统中所需的最有效且安全的药物。
[0129]
对于氰化物中毒,通过静脉注射给予患者剂量至多12.5g的sts。当用于降低顺铂的肾毒性时,在顺铂之前给患者静脉内给予第一剂4g/m2体表面积的sts,然后与顺铂同时给予剂量通常为12g/m2的第二剂。在顺铂外渗的情况下,每100ml顺铂要静脉注射0.167m的sts溶液2ml,然后在外渗区域周围以顺时针方向皮下注射0.1ml,至多1ml。该过程在外渗事件发生后3-4小时内重复数次。为了治疗与顺铂相关的耳毒性,在耳中施用含0.1m sts溶液的透明质酸凝胶。在花斑癣的治疗中,每天两次经皮施用sts的15%的乳液配方,持续4周。
[0130]
据报道,静脉注射sts(37.5-75.0g/周)可改善受钙化防御影响的透析患者的皮肤损伤。此外,尽管sts的经口摄取量较低,但有多个研究描述了此类患者成功经口使用高达7.5g/周的sts(musso等人,saudi journal of kidney diseases and transplantation 2008,19,820-821;shetty和klein,advances in peritoneal dialysis 2016,32,51-55)。
[0131]
sts也经常用作药物制剂(包括眼科制剂)中的赋形剂。
[0132]
adc通常通过静脉内输注施用。adc与硫代硫酸盐“组合”施用或反之亦然,在本文中应理解为表示adc和硫代硫酸盐作为相同治疗方案的组分施用,其中施用硫代硫酸盐以预防或治疗非期望的非靶组织毒性。然而,不要求硫代硫酸盐和adc包含在单一药物制剂中。在一个优选的实施方案中,adc和硫代硫酸盐同时、分开或依次施用。合适的剂量和剂型的确定尤其取决于要预防或治疗的非期望的非靶组织毒性,并且由临床医生确定,例如,使用本领域已知的或疑似的影响治疗或预测影响治疗的参数或因素。治疗方案可以是例如包括在adc首次施用前约三周至施用后约1小时中首次施用硫代硫酸盐,以及随后以规律的间隔施用硫代硫酸盐直至adc最后一次施用之后最多三个月。这样的规律间隔可以是例如包括但不限于每天三次、每天两次、每天、每周、每两周一次。
[0133]
在进一步的实施方案中,硫代硫酸盐通过吸入或者通过静脉内、经口、经皮肤、皮下或眼部途径施用。
[0134]
在一个优选的实施方案中,硫代硫酸盐通过静脉内途径施用。在一个更优选的实施方案中,硫代硫酸盐通过静脉内途径施用,随后通过皮下途径施用,这可用于治疗由adc外渗引起的毒性。通常,在外渗后,静脉内注射有效量的硫代硫酸盐溶液,然后在外渗区域周围多次皮下注射有效量的硫代硫酸盐溶液。通常,对每100ml的adc要静脉注射多达10ml
的0.05-0.5m硫代硫酸盐溶液,然后在外渗区域周围以顺时针方向皮下注射0.1ml,至多1ml。在外渗事件发生后3-12小时内,优选3-8小时内,更优选3-6小时内,最优选3-4小时内,重复该过程数次。
[0135]
在另一个优选的实施方案中,硫代硫酸盐通过眼部途径施用。通常,有效量的硫代硫酸盐以滴眼剂的形式施用。通常,每只眼睛施用1或2滴的0.0313-0.5m硫代硫酸盐溶液,每天施用1至24次。
[0136]
在另一方面,本发明提供一种包含硫代硫酸盐的组合物,优选其中所述组合物是药物组合物,更优选其进一步包含药学上可接受的载体。这种组合物在下文中称为本发明所述的组合物。所述组合物可以是例如液体制剂、冻干制剂或例如胶囊或片剂的形式。
[0137]
优选的形式取决于预期的施用方式和治疗应用。药用载体可以是适合将硫代硫酸盐递送至受试者的任何相容的、无毒的物质。药学上可接受的载体是本领域公知的并且包括例如一种或更多种水溶液如(无菌)水或生理缓冲盐水或溶剂或载体如乙二醇、甘油、透明质酸、油如橄榄油或可注射的有机酯、酒精、脂肪、蜡和惰性固体。药学上可接受的载体可以进一步包含生理学上可接受的化合物,其起到例如稳定或提高硫代硫酸盐吸收的作用。这种生理上可接受的化合物包括例如一种或更多种碳水化合物如葡萄糖、蔗糖或葡聚糖,抗氧化剂如抗坏血酸或谷胱甘肽,螯合剂,低分子量蛋白质,或另一种稳定剂或赋形剂。本领域技术人员知道药学上可接受的载体(包括生理学上可接受的化合物)的选择取决于例如组合物的施用途径。本发明的药物组合物可以进一步包含一种或更多种药学上可接受的佐剂、缓冲剂(如柠檬酸盐、氨基酸如组氨酸或含琥珀酸盐的水盐)、冻干保护剂(如蔗糖、海藻糖)、张力调节剂(如氯化物盐例如氯化钠)、表面活性剂(如聚山梨醇酯)、填充剂(如甘露醇、甘氨酸)等。
[0138]
对于经口施用,硫代硫酸盐可以以固体剂型如胶囊、片剂和粉剂施用,或以液体剂型如酏剂、糖浆剂或混悬剂施用。例如,硫代硫酸盐可以与非活性成分和粉末状载体(如葡萄糖、乳糖、蔗糖、甘露醇、淀粉、纤维素或纤维素衍生物、硬脂酸镁、硬脂酸、糖精钠、滑石粉、碳酸镁等)一起封装在明胶胶囊中。可添加以提供所需颜色、味道、稳定性、缓冲能力、分散性或其他已知所需特征的额外的非活性成分的实例是氧化铁红、硅胶、十二烷基硫酸钠、二氧化钛、可食用白色墨水等。类似的稀释剂可用于制备压缩片剂。片剂和胶囊都可以制成缓释产品,以在数小时内连续释放硫代硫酸盐。压缩片剂可以是糖衣或薄膜包衣以掩盖任何令人不快的味道并保护片剂免受大气影响,或可以是肠溶衣以在胃肠道中选择性崩解。用于经口施用的液体剂型可以包含着色剂和矫味剂以提高患者的接受度。
[0139]
用于肠胃外施用的制剂必须是无菌的。灭菌很容易通过任选地在冻干和重构之前或之后用无菌过滤膜过滤来完成。用于施用的肠胃外途径与已知方法一致,例如通过静脉内、腹膜内、肌肉内、动脉内或病灶内途径注射或输注。所述组合物可以通过输注或快速推注连续施用。用于静脉内输注的典型组合物可包含100至500ml无菌0.9%nacl或5%葡萄糖,任选地补充有20%白蛋白溶液和1mg至10g的活性化合物,这取决于特定类型的化合物及其所需的施用方案。制备肠胃外施用组合物的方法在本领域中是众所周知的并且在多种来源中都有更详细的描述,包括例如remington's pharmaceutical science(第17版,mack publishing,easton,pa,1985)。
[0140]
本发明所述的组合物可以是速释组合物或是可延迟、改良或持续释放的组合物。
[0141]
在一个实施方案中,本发明涉及一种药物组合物,其包含高达1.0m的sts和透明质酸,其中所述组合物是适用于眼部使用的液体组合物。优选地,所述组合物包含至多0.5m的sts,例如0.0313、0.05、0.063、0.10、0.125、0.20、0.25或0.5m。
[0142]
在另一个实施方案中,本发明涉及一种药物组合物,其包含高达250mg/ml的sts和至少一种药学上可接受的载体,其中所述组合物是适合静脉内使用的液体组合物。合适的药物载体是例如注射用水、盐水或氯化钾溶液,以及用于ph调节的氢氧化钠或硼酸。
[0143]
在另一个实施方案中,本发明涉及一种药物组合物,其包含高达25%的sts和至少一种药学上可接受的载体,其中所述组合物是适合皮肤使用的液体组合物。合适的药物载体是例如注射用水、异丙醇和丙二醇。
[0144]
在另一方面,本发明提供了一种包含硫代硫酸盐的组合物,其用于哺乳动物,优选人,以预防或降低向哺乳动物施用如上文所定义的adc相关的非靶组织毒性。
[0145]
在另一方面,本发明提供了如上文所定义的adc在制备用于治疗哺乳动物,优选人的肿瘤的组合疗法的药物中的用途,其中所述药物与如上文定义的硫代硫酸盐组合施用。
[0146]
在另一方面,本发明提供了一种含有如上文所定义的adc和硫代硫酸盐作为组合制剂的产品,以用于同时、分开或依次治疗哺乳动物,优选人的肿瘤。本文中所用的“组合制剂”特别地定义“成套制剂(kit of parts)”,意思是如上文定义的组合伴侣可以独立施用或通过使用具有不同量组合伴侣的不同固定组合施用,即同时、分开或依次施用。在一些实施方案中,成套制剂的各部分可以例如同时或按时间交错施用,即成套制剂的任何部分都可以在不同的时间点并间以相同或不同的时间间隔施用。在一些实施方案中,组合伴侣总量的比例可以在组合制剂中施用。所述产品优选是药物产品。所述产品可以包含在容器、包装或分配器中,可选地与施用说明一起提供。
[0147]
在另一方面,本发明提供了一种预防或降低与施用如上文定义的adc相关的非靶组织毒性的方法,包括组合施用有效量的adc与有效量的硫代硫酸盐。在一个实施方案中,硫代硫酸盐在adc第一次施用前约三周至施用后约1小时内施用,并且硫代硫酸盐的施用以规律的间隔重复,直到adc治疗结束之后最多三个月,即最多至最后一次adc施用后三个月。这种规律的间隔可以是例如包括但不限于每天三次、每天两次、每天、每周、每两周一次。在一个优选的实施方案中,硫代硫酸盐在adc第一次施用之后施用。在另一个优选的实施方案中,adc在硫代硫酸盐第一次施用之后施用。
[0148]
在另一方面,本发明涉及一种治疗、预防或降低与施用如上文所定义的adc相关的非靶标毒性的方法,所述方法包括向有需要的受试者施用治疗有效量的硫代硫酸盐。本文中所用的术语“受试者”指归类为哺乳动物的所有动物,包括但不限于灵长类动物和人。受试者优选是人。“治疗有效量”的表述是指有效治疗、预防或降低与施用adc相关的非期望毒性的量;所述量可以是足以实现所需反应或改善症状或体征的量。特定受试者的治疗有效量可根据诸如所治疗的病症、受试者的整体健康状况、施用的方法、途径和剂量以及副作用的严重程度等因素而变化。
[0149]
在本文件及其权利要求书中,动词“包含”及其变位以其非限制性意义使用,表示包括该词之后的项目,但不排除未具体提及的项目。此外,没有数量词修饰的名词表示一个/种或更多个/种,除非上下文明确要求存在一个且只有一个要素。因此,没有数量词修饰的名词通常意指“至少一个/种”。
[0150]
本说明书中引用的所有专利和参考文献的全部内容通过引用并入本文。
[0151]
以下实施例仅用于说明目的,并不旨在以任何方式限制本发明的范围。
[0152]
实施例
[0153]
材料和方法
[0154]
lc-ms-对于lc-ms,将10μl样品以0.11ml/min的流速注入acquity uplc beh shield rp18色谱柱(粒径1.7μm,1.0mm id x 100mm,waters,货号1270),柱温为45℃。洗脱方法如下表所示。流动相a的组成为含0.1%甲酸的milli-q水,流动相b为乙腈。运行时间为9.0分钟。使用配备有microtof q ii质谱仪(bruker)、empower软件(uplc)以及bruker软件(ms q-tof)的waters acquity uplc系统。在275-279nm处测量uv吸光度。
[0155]
梯度程序
[0156][0157]
实施例1
–
硫醇化合物的解毒实验
[0158]
含环丙基(倍癌霉素)化合物syd986的实验
[0159][0160]
将syd986在含0.1%乙酸的二甲基乙酰胺(dimethylacetamide,dma)中溶解至5.0μg/ml(10μm)的浓度。将下表中列出的硫醇溶解在乙腈/水(1:1)中。
seco-duba)例如在氯原子处反应,将硫代硫酸盐添加至(n-乙酰半胱氨酸-)淬灭的vc-seco-duba。使用淬灭的接头-药物代替vc-seco-duba以防止硫代硫酸盐与接头-药物的马来酰亚胺部分发生反应。
[0169][0170]
用含0.1%乙酸的dma将淬灭的接头-药物溶解并稀释到15.0μg/ml(10μm)的浓度。将sts溶解在乙腈/水(1:1)中。
[0171]
通过将900μl的11.1mm的sts溶液与100μl的淬灭接头-药物溶液(淬灭接头-药物1μm;sts 10mm)混合来制备解毒测试溶液。在实验开始时和不同时间间隔后进行lc-ms分析。
[0172]
在24小时的测试时间范围内,淬灭的接头-药物和硫代硫酸盐之间没有发生反应。由于硫代硫酸盐不与淬灭的接头-药物反应,因此包含该接头-药物的vc-seco-duba和/或adc不太可能在体内受到影响。
[0173]
pbs缓冲液中的实验
[0174]
用syd986和淬灭的接头-药物在磷酸盐缓冲盐水(phosphate buffered saline,pbs)(1x)缓冲液中进行了类似的实验。syd986用0.5μg/ml和0.05μg/ml(1.0和0.1μm)进行测试。淬灭的接头-药物用1.5μg/ml和0.15μg/ml(1.0和0.1μm)进行测试。
[0175]
1xpbs的最终浓度为137mm nacl、10mm磷酸盐、2.7mm kcl,且ph值为7.4。
[0176]
pbs中的结果与上述乙腈/水(1:1)中的结果相当。
[0177]
实施例2
–
sts的体外解毒实验
[0178]
sk-br-3细胞中的细胞活力测试
[0179]
在存在或不存在1mm sts的情况下,将sk-br-3细胞在含环丙基化合物syd986或adc syd1875中暴露6天。syd1875是一种adc,包含抗5t4抗体和接头-药物vc-seco-duba(clinicaltrials.gov nct04202705)。
[0180]
将完全生长培养基中的sk-br-3细胞以每孔6500个细胞(90μl/孔)接种在96孔板中,并在37℃、5%co2下孵育。过夜孵育后,加入10μl含有或不含sts的syd986或syd1875。在培养基中进行系列稀释。在固定的1mm sts浓度实验中,制备稀释范围为syd986或syd1875浓度的预混合物,并在将其添加至固定浓度的sts中之后再添加至细胞。
[0181]
根据制造商的说明,在6天后使用来自invitrogen的prestoblue
tm
细胞活力测定法和cyquant
tm
直接细胞增殖测定法来评估细胞活力。通过将在存在或不存在sts的情况下每
个syd986或syd1875浓度下测得的荧光除以未处理的细胞的平均平均值乘以100来计算存活百分比。未处理的细胞仅暴露于适当的载体,即syd986(含或不含sts)的实验中使用含有1%的生长培养基dmso 0.25%h2o,在有sts的情况下,syd1875的实验中使用含有0.25%h2o的生长培养基,而在没有sts的情况下,syd1875的实验中仅使用生长培养基。
[0182]
如图4a所示,在sk-br-3细胞中,1mm sts对syd986的体外效力产生负面影响,因此表明sts可以对活性倍癌霉素药物进行解毒。且有4.6倍的效力转变。
[0183]
如图4b所示,在sk-br-3细胞中,1mm sts对抗5t4 adc syd1875的体外效力产生负面影响,因此表明sts可以在细胞内释放活性倍癌霉素药物后对其进行解毒。且有27倍的效力转变。
[0184]
实施例3-通过眼部途径施用sts的体内安全性实验
[0185]
用浓度高达0.3m sts的配方在新西兰白兔中进行的局部耐受性和毒性研究没有显示出不良发现。
[0186]
下面概述了确认sts在人眼睛中具有良好耐受性的合适方案:
[0187]
可以要求健康受试者在双眼中自我施用包含硫代硫酸盐的滴眼剂14天。每位受试者在第1天施用一次滴眼剂,在第2天施用3次(从早晨开始,每2-3小时一次),在随后的12天中每天施用6次(在清醒时每2-3小时一次)。每次施用受试者应在每只眼睛中施用一滴。可以使用0.02、0.05、0.10和0.20m sts的浓度,配方如表1至4中详述。
[0188]
表1.包含0.02m sts的组合物
[0189][0190]
q.s.=足量(quantum satis)
[0191]
表2.包含0.05m sts的组合物
[0192][0193]
q.s.=足量(quantum satis)
[0194]
表3.包含0.10m sts的组合物
[0195]
[0196][0197]
q.s.=足量(quantum satis)
[0198]
表4.包含0.20m sts的组合物
[0199][0200]
q.s.=足量(quantum satis)
[0201]
实施例4-通过眼部途径施用sts以降低包含倍癌霉素衍生物的抗体-药物缀合物的潜在非靶眼部毒性的方案
[0202]
以下方案说明了sts可以在临床环境中使用的方式。通过眼部途径施用sts(例如作为滴眼剂),可以降低包含倍癌霉素衍生物的抗体-药物缀合物的潜在非靶眼部毒性。
[0203]
含有sts的滴眼剂(如实施例3中表3和4中所例示的具有0.10m或0.20m sts的滴眼制剂)可以在syd985(vc-seco-duba与抗her2抗体曲妥珠单抗的adc)治疗的同时(自我)施用。
[0204]
当syd985以每三周1.2mg/kg体重的剂量施用时,从第一次输注syd985之日起至最后一次输注之后21天或直至决定停止syd985治疗(以较晚者为准)期间,sts滴眼剂每天最
多可使用6次(清醒时大约每2至3小时一次)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。